Хроматография (ОФС.1.2.1.2.0001.15). Методы, параметры
» data-shape=»round» data-use-links data-color-scheme=»normal» data-direction=»horizontal» data-services=»messenger,vkontakte,facebook,odnoklassniki,telegram,twitter,viber,whatsapp,moimir,lj,blogger»>
Хроматография (ОФС.1.2.1.2.0001.15)
Хроматография – метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения.
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Взамен ст. ГФ XI, вып.1
Хроматографией называется метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения. По механизму, лежащему в основе разделения, различают адсорбционную, распределительную, ионообменную и другие виды хроматографии.
В настоящее время используются следующие хроматографические методы анализа, представленные на рис.1
Рисунок 1. Методы хроматографического анализа.

Результат хроматографического разделения представляется в виде хроматограммы.
ХРОМАТОГРАММА И ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
Хроматограмма представляет собой графическое или иное представление сигнала детектора, концентрации веществ в элюате или другой количественной величины, используемой для измерения концентрации веществ в элюате, от времени или объема подвижной фазы. В планарной (плоскостной) хроматографии хроматограммой называют также зафиксированную на бумаге (бумажная хроматография) или ТСХ-пластинке (тонкослойная хроматография) последовательность зон адсорбции веществ исходной (анализируемой) смеси.
Схематически хроматограммы представляют собой последовательность гауссовых пиков на базовой линии (рис. 2).
Базовая линия – сигнал от подвижной фазы.
Пик – часть хроматограммы, регистрирующая отклик детектора. Пик отображает постепенное нарастание концентрации вещества и последующее ее уменьшение. В случае линейной изотермы сорбции кривая, описывающая пик, приближается к кривой гауссова распределения.
Основание пика — продолжение базовой линии, соединяющее начало и конец пика.
Площадь пика (S) – площадь хроматограммы, заключенная между кривой, описывающей пик, и его основанием.
Высота пика (H) – расстояние от максимума пика до его основания, измеренное параллельно оси отклика детектора.

Рисунок 2. Хроматограмма и основные хроматографические параметры: 1 и 2 – пики соединений 1 и 2; t1 и t2 – соответствующие времена удерживания; t0 – время удерживания несорбирующегося вещества; W1 и W2 – ширина пиков у основания; W0,5 – ширина пика на половине его высоты (предполагается гауссова форма пиков)
Интерпретация хроматографических данных
Время удерживания (tR или t) – время, необходимое для элюирования вещества. Соответствует времени появления максимума пика на хроматограмме.
Объем удерживания (VR) – объем подвижной фазы, необходимый для элюирования вещества. Может быть вычислен по времени удерживания и скорости потока (F).
Объем удерживания, в отличие от времени удерживания, не зависит от скорости потока.
Время удерживания несорбирующегося вещества (t0 или tм) – время, необходимое для элюирования неудерживаемого на сорбенте вещества. В эксклюзионной хроматографии t0 соответствует времени удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Объем удерживания несорбирующегося вещества (V0) – объем подвижной фазы, необходимый для элюирования неудерживаемого вещества. Может быть вычислен по времени удерживания неудерживаемого вещества и скорости потока (F):
В эксклюзионной хроматографии V0 соответствует объему удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Общее время удерживания подвижной фазы (tt) – в эксклюзионной хроматографии время удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Общий объем удерживания подвижной фазы (Vt) – в эксклюзионной хроматографии объем удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Константа (коэффициент) распределения (K0) – в эксклюзионной хроматографии характеристика элюирования вещества из определенной колонки, которую рассчитывают с помощью выражения:

Приведенное (исправленное) время удерживания вещества (t´R) ‒ время удерживания вещества за вычетом времени удерживания несорбируемого вещества. Может быть рассчитано по формуле:
Исправленное время удерживания не зависит от объема трубопроводов хроматографической системы, установленных между инжектором и колонкой.
Относительное время удерживания (r) – относительное приведенное (исправленное) время удерживания вещества 2 по веществу 1:

Нескорректированное относительное время удерживания (RRT) – относительное время удерживания вещества 2 по веществу 1:

Если не указано иное, значения относительного времени удерживания, приведенные в фармакопейных статьях, соответствуют нескорректированному относительному времени удерживания
Коэффициент емкости (k´) – коэффициент емкости колонки по веществу с временем удерживания tR, показывающий, во сколько раз исправленное время удерживания вещества больше, чем время удерживания несорбируемого вещества.

Эффективность хроматографической системы ‒ параметр, характеризующий степень размывания хроматографического пика. Эффективность выражается числом теоретических тарелок (N):

W – ширина пика у основания;
W0,5 – ширина пика на половине высоты.
При расчете числа теоретических тарелок значения времени удерживания и ширины пика должны быть приведены к одинаковой размерности.
Число теоретических тарелок зависит от природы определяемого вещества, его концентрации или объема, вводимого в систему, от колонки, температуры колонки и состава подвижной фазы.
Если не указано иное, то эффективность хроматографической системы, требования к которой приведено в фармакопейной статье, рассчитывается по формуле, использующей ширину пика на половине его высоты.
Фактор асимметрии (фактор симметрии) пика (As) рассчитывают по формуле:
 где
где
W0,05 – ширина пика на 5 % (1/20) его высоты;
f – расстояние между перпендикуляром, опущенным из вершины пика, и восходящей стороной пика на 5 % его высоты (рис. 3).
Если фактор асимметрии равен 1, то пик симметричен. Если фактор асимметрии больше 1, то это означает, что растянут задний фронт пика. Если фактор асимметрии меньше 1, то пик растянут спереди.

Рисунок 3. Схема расчета фактора асимметрии пика
Разрешение (Rs).
Разрешение между пиками двух веществ смеси элюирующимися друг за другом рассчитывают по формулам:

при этом tR2 ≥ tR1. При расчете разрешения величины времени удерживания и ширины пиков должны быть приведены к одинаковой размерности.
Если не указано иное, то разрешение, требования к которому приведены в фармакопейной статье, рассчитывается по формуле, использующей ширину пиков на половине высоты.
В случае, если пики несимметричны и если интенсивность пиков значительно различается, параметр Rs не всегда корректно описывает разделение хроматографических пиков. Таким образом, даже при значениях Rs ≥ 1,5 может наблюдаться неполное разделение пиков. В этих случаях при оценке разделяющей способности можно заменить параметр Rs на параметр «отношение максимум/минимум».
Отношение максимум/минимум (p/v), называемое также отношением «peak-to-valley», «пик – долина». Этот параметр позволяет оценить разделительную способность хроматографической системы. Значение p/v рассчитывается по формуле:
Hp – высота меньшего пика относительно экстраполированной базовой линии;
Hv – высота низшей точки (седловины) кривой, разделяющей пики, относительно экстраполированной базовой линии (рис. 4).

Рисунок 4. Хроматограмма не полностью разделяемых веществ
Данное соотношение применяется для оценки разделительной способности хроматографической системы, если вещества смеси разделяются не полностью. Рассчитанное отношение максимум/минимум в значительной степени зависит от выбранного варианта интегрирования хроматограммы. Результаты измерения соотношения p/v будут некорректны в случае разметки меньшего пика методом экстраполяции смещенной базовой линии (методом тангенциальной касательной).
Отношение сигнал/шум (S/N).
Краткосрочный шум сигнала детектора (шум базовой линии) влияет на прецизионность количественного определения. Отношение сигнал/шум рассчитывают по формуле:

Н – высота пика, соответствующего рассматриваемому веществу на хроматограмме указываемого стандартного раствора, измеренная от максимума пика до экстраполированной базовой линии. Экстраполяция базовой линии проводится для сигнала на участке базовой линии во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте;
h – размах фонового шума, измеряемый либо на хроматограмме контрольного (холостого) раствора (или раствора плацебо), либо на хроматограмме того же раствора стандартного образца.
Измерение размаха фонового шума проводится во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте, расположенном, если это возможно, равномерно по обе стороны от места возможного обнаружения пика. В случае использования для измерения шума хроматограммы контрольного (холостого) раствора (или раствора плацебо) измерение размаха фонового шума проводится во временном интервале, включающем в себя время удерживания рассматриваемого вещества, при этом продолжительность временного интервала, в котором проводится измерение шума, должна не менее чем в 5 раз превышать ширину на половине высоты для пика на хроматограмме указываемого раствора стандартного образца (рис. 5).

Рисунок 5. Вычисление отношения сигнал/шум.
Объем задержки (D) (объем задержки градиента) представляет собой объем системы между точкой, в которой происходит смешение элюентов, и началом колонки.
Объем задержки градиента влияет на времена удерживания веществ и общий профиль наблюдаемой хроматографической картины, получаемой при использовании градиентного элюирования. Объем задержки хроматографической системы определяется следующим способом.
Колонка. Заменяют хроматографическую колонку капилляром (например, 1 м × 0,12 мм).
Подвижная фаза.
Подвижная фаза A — вода.
Подвижная фаза B — 0,1 % (об/об) раствор ацетона.
Скорость потока. Необходимая для создания давления, достаточного для стабильной работы насоса (например, 2 мл/мин).

Детектор. Спектрофотометрический при длине волны 265 нм.
Определяют время (t0,5) в минутах, когда оптическая плотность увеличилась на 50 %. Вычисляют объем задержки:


Рисунок 6. Определение объема задержки градиента
Прецизионность системы в условиях повторяемости
Прецизионность системы в условиях повторяемости выражается в виде рассчитанного относительного стандартного отклонения в процентах [RSD (%)] по результатам последовательных измерений не менее чем 3 вколов или нанесений раствора сравнения и рассчитывается по формуле:

В планарной хроматографии аналогом времени удерживания является фактор удерживания (Rf):
Rf = a / b
где a – расстояние от точки нанесения пробы до центра пятна, характеризующего зону адсорбции;
b – расстояние от линии старта до линии фронта элюента.
На экспериментально определяемые значения Rf заметно влияют условия хроматографирования. Оценкой хроматографической подвижности, менее чувствительной к влиянию отклонений в условиях проведения эксперимента, является величина Rst, представляющая собой отношение величины Rf одного вещества к величине Rf другого, принятого за стандарт:


Рисунок 7. Схема определения значений Rf и Rst.
Данные планарной хроматографии могут быть представлены в виде денситограмм.
РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ ВЕЩЕСТВ
При расчетах содержания определяемых веществ пики растворителей и реактивов, подвижной фазы или среды (матрицы) образца не учитываются.
Существуют 4 основных метода расчета концентрации анализируемого вещества по хроматографическим данным.
1. Метод нормирования (метод внутренней нормализации). Применение данного метода основано на предположении, что на хроматограмме зарегистрированы все вещества, входящие в состав анализируемой смеси, и что доля площади (высоты) каждого пика от суммы площадей (высот) всех пиков соответствует содержанию вещества в массовых процентах. Процентное содержание вещества в анализируемой смеси рассчитывается путём определения площади соответствующего пика как процентной части общей площади всех пиков, за исключением пиков, соответствующих растворителям или реактивам, подвижной фазе или матрице образца. Содержание каждого вещества в смеси в процентах может быть вычислено по формуле:
Если чувствительность детектора различна по отношению к каждому из веществ, то вводят поправочные коэффициенты ki. Относительный коэффициент отклика детектора, обычно называемый фактором отклика, обозначает чувствительность детектора для данного вещества относительно стандартного вещества. Поправочный коэффициент – это число, обратное фактору отклика.

Поправочные коэффициенты рассчитывают относительно основного вещества анализируемой смеси или другого стандартного вещества по формуле:
 где
где
Сi и С0 – концентрация i-го вещества и стандартного вещества соответственно;
Si и S0 – площадь (высота) пика i-го вещества и стандартного вещества соответственно.
Данные коэффициенты могут не учитываться в случае, если они находятся в пределах диапазона 0,8 – 1,2.
При использовании поправочных коэффициентов выражение для расчета количественного содержания приобретает вид:

При проведении испытания на примеси методом нормализации или методом внешнего стандарта с использованием разведения раствора испытуемого образца в качестве раствора сравнения учитывают все указанные в нормативной документации поправочные коэффициенты, значение которых выходит за пределы диапазона 0,8 – 1,2.
2. Метод внешнего стандарта. Концентрацию испытуемого вещества определяют путём сравнения сигнала (пика), полученного на хроматограммах испытуемого раствора, и сигнала (пика), полученного на хроматограммах раствора стандартного образца.
Концентрацию определяемого вещества в испытуемом растворе рассчитывают по формуле:

S и S0 – средние значения площадей (высот) пиков на хроматограммах испытуемого и стандартного растворов соответственно;
С и С0 – концентрации определяемого и стандартного растворов соответственно.
Количественное определение содержания примесей методом внешнего стандарта предпочтительнее проводить с использованием стандартных растворов примесей с концентрациями, близкими к их ожидаемым концентрациям в испытуемом растворе.
В качестве раствора стандартного образца для количественного определения примесей возможно использование раствора основного вещества. В этом случае разведение подбирается таким образом, чтобы концентрация основного соединения в растворе стандартного образца по отношению к его концентрации в испытуемом растворе была близка к ожидаемой концентрации примесей в испытуемом растворе. В этом случае следует учесть факторы отклика примесей по отношению к основному веществу, если их значения выходят за рамки 0,8 — 1,2.
Частным случаем метода внешнего стандарта является метод калибровочной кривой, в ходе которого определяют взаимосвязь между измеренным или обработанным сигналом (у) и количеством (концентрацией, массой и т.д.) определяемого вещества (х) и рассчитывают уравнение калибровочной функции. Результаты испытания рассчитывают из измеренного или обработанного сигнала с помощью обратной функции.
3. Метод внутреннего стандарта. Концентрацию определяемого вещества определяют путём сравнения отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме испытуемого раствора и отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме раствора стандартного образца. Метод внутреннего стандарта основан на введении в анализируемую смесь определенного количества стандартного вещества (внутренний стандарт). В испытуемый и стандартный растворы вводят известные количества внутреннего стандарта, хроматографируют растворы и определяют отношения площадей (высот) пиков определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом и стандартном растворах.
Концентрацию определяемого вещества (Х) рассчитывают по формуле:

4. Метод стандартных добавок. В качестве внутреннего стандарта выбирается вещество, отсутствующее в испытуемой пробе, не взаимодействующее с определяемым веществом и другими веществами пробы, обладающее достаточной стабильностью, полностью отделяющееся от веществ пробы и не содержащее примесей с временами удерживания, совпадающими с временем удерживания определяемого вещества. Концентрация внутреннего стандарта должна быть близка к концентрации определяемых веществ, а структура и свойства по возможности аналогичны структуре и свойствам определяемых веществ.
Концентрация определяемого вещества определяется путём сравнения сигнала (площади или высоты пика), соответствующего определяемому веществу, на хроматограмме испытуемого раствора, и сигнала (площади или высоты пика) определяемого вещества на хроматограмме испытуемого раствора с известной добавкой определяемого вещества. Метод стандартных добавок основан на введении в анализируемую смесь известного количества определяемого вещества и сравнения сигналов, полученных для испытуемого раствора со стандартной добавкой и без добавки определяемого вещества. При внесении стандартной добавки стараются минимизировать разбавление испытуемого образца, чтобы измерения раствора со стандартной добавкой и без проходили в одинаковых условиях с одинаковым влиянием матрицы. Количество вводимого в стандартной добавке определяемого вещества должно быть соизмеримо с его предполагаемым содержанием в испытуемом образце. После проведения испытания сравнивают полученные значения интенсивности и рассчитывают количественное содержание определяемого вещества Сх по формуле:

Сстд — концентрация стандартной добавки;
Sx — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора без стандартной добавки;
Sстд+х — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора со стандартной добавкой.
При необходимости формулу корректируют с учетом разбавлений испытуемого раствора за счет введения стандартной добавки.
РЕКОМЕНДАЦИИ ПО РАЗМЕТКЕ И ИНТЕГРИРОВАНИЮ ХРОМАТОГРАММ ПРИ ОПРЕДЕЛЕНИИ ПРИМЕСЕЙ
Если не указано иное, в испытаниях на содержание примесей в качестве порога игнорирования (неучитываемый предел, уровень содержания вещества, при котором и при меньшем содержании пик не принимается во внимание) принимают величину 0,05 % от содержания основного вещества.
В случаях, когда при испытании на примеси требуется определение суммарного содержания примесей или количественного определения примеси, при интерпретации хроматограммы важно выбрать подходящие условия интегрирования и значения порога интегрирования [интенсивность сигнала пика соединения (высоты или площади)], при котором (и при меньшем) пик не учитывается системой сбора данных и не участвует в количественных расчетах.
С целью разметки на хроматограмме всех пиков, подлежащих учету, заданный порог интегрирования системы сбора данных не должен превышать половину порога игнорирования.
Интегрирование площади пика любой примеси, пик которой не полностью разделяется с основным пиком, предпочтительно проводить экстраполяцией смещенной базовой линии (методом тангенциальной касательной). Использование других способов интегрирования должно быть специально оговорено в фармакопейной статье.
ОЦЕНКА ПРИГОДНОСТИ ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ
Испытания пригодности системы являются неотъемлемой частью методики и используются для того, чтобы убедиться в надлежащем функционировании хроматографической системы и обеспечить выполнение предъявляемых к ней требований.
При проведении испытаний используемое оборудование должно быть квалифицировано и способно к функционированию надлежащим образом.
Для оценки пригодности системы указывают:
- требования к параметрам, характеризующим форму пика [эффективность хроматографической системы N и фактор асимметрии (фактор симметрии) As];
- требования к разделительной способности (разрешение между пиками RS или отношение пик — долина p/v);
- требования к воспроизводимости (относительное стандартное отклонение, RSD) значений площади или высоты пиков, а также времен удерживания пиков в случае оценки подлинности соединений;
- требования к чувствительности при проведении испытания на примеси [минимально определяемая концентрация (фактический предел количественного определения) примеси]. Оценивается посредством расчета отношения сигнал/шум для раствора соответствующей концентрации.
Если в фармакопейной статье не указано иное, должны выполняться следующие требования и все дополнительные требования, приведенные в нормативном документе:
- в испытаниях на примеси или количественное содержание для пика на хроматограмме растворе стандартного образца, используемого для количественных определений, значение величины фактора асимметрии (фактора симметрии) As должно находиться в пределах от 0,8 до 1,5;
- разрешение между пиками Rs ³ 1,5 (в случае не полностью разделенных пиков вместо разрешения может быть использовано соотношение p/v. При этом требования к минимальному значению соотношения p/v устанавливаются в фармакопейной статье);
- отношение сигнал/шум для пика вещества, полученное для раствора с концентрацией, равной требуемому уровню минимально определяемой концентрации, должно быть не менее 10. Требуемый уровень минимально определяемой концентрации [фактический предел количественного определения (ПКО)] зависит от того, предполагает ли методика вычисление содержания примесей, или только полуколичественную оценку, когда результат представляется в виде «менее Х» или «не более Х», где Х – допустимое содержание примеси. Для методик, предполагающих вычисление содержания примесей, минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать значение порога игнорирования (если не указано иное – 0,05 % относительно концентрации основного вещества в испытуемом растворе). Для полуколичественных методик минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать максимально допустимое содержание примеси. При необходимости оценки содержания нескольких примесей требуемый уровень минимальной определяемой концентрация для применяемой хроматографической системы определяется примесью, нормы содержания которой наиболее строги. Если в фармакопейной статье не указано иное, то раствор вещества минимально определяемой концентрации для оценки чувствительности детектирования можно приготовить растворением стандартного образца вещества в том же растворителе, который используется для приготовления испытуемого раствора, с уровнем концентрации 0,05 % относительно концентрации основного вещества в испытуемом растворе.
Требования к прецизионности системы в условиях повторяемости при количественном определении основного вещества в субстанциях
При количественном определении основного вещества в фармацевтических субстанциях при содержании основного вещества, близком к 100 %, максимально допустимое относительное стандартное отклонение значений интенсивности (площади или высоты) пика основного вещества для заданного количества повторных введений стандартного раствора RSDmax вычисляется по формуле:
Если в фармакопейной статье не указано иное, то значение RSD не должно превышать величин RSDmax, приведенных в таблице. Данное требование не применяется при испытаниях на примеси.

Таблица. Максимально допустимое относительное стандартное отклонение RSDmax в зависимости от верхнего предела содержания основного вещества и числа отдельных введений проб
| В, % | Число отдельных введений проб (вколов) | |||
| 3 | 4 | 5 | 6 | |
| Максимально допустимое относительное стандартное отклонение RSDmax | ||||
| 2,0 | 0,41 | 0,59 | 0,73 | 0,85 |
| 2,5 | 0,52 | 0,74 | 0,92 | 1,06 |
| 3,0 | 0,62 | 0,89 | 1,10 | 1,27 |
Соответствие критериям пригодности системы должно поддерживаться на протяжении всего испытания. В зависимости от различных факторов, например, частоты использования методики и опыта работы с хроматографической системой, аналитик выбирает соответствующую схему проверки, подтверждающую соответствие этим требованиям.
В планарной хроматографии при проведении испытаний, регламентирующих наличие посторонних примесей/родственных соединений, должно быть предусмотрено определение предела обнаружения определяемых примесей и разделительной способности хроматографической системы.
Для подтверждения пригодности системы возможно одновременное использование двух тестов: «Подтверждение разделительной способности» и «Подтверждение чувствительности».
Тест «Подтверждение разделительной способности» проводят путём нанесения на пластинку и последующего хроматографирования специального стандартного раствора, содержащего два или более веществ с известными значениями Rf. После хроматографирования эти вещества должны разделиться, образуя зоны адсорбции, значения Rf которых равны заданным.
Тест «Подтверждение чувствительности» позволяет оценить чувствительность проявления. Он заключается в нанесении определённого количества стандартного вещества, хроматографируемого в условиях предыдущего теста и проявляемого тем же реагентом. Зона адсорбции стандартного вещества должно чётко обнаруживаться.
В каждом случае природа стандартных веществ, состав стандартного раствора, наносимые количества, приблизительные значения Rf устанавливают в фармакопейных статьях.
При необходимости для достижения требуемых критериев пригодности хроматографической системы проводится корректировка хроматографических условий.
КОРРЕКТИРОВКА УСЛОВИЙ ХРОМАТОГРАФИРОВАНИЯ
Далее приводятся пределы возможных изменений различных параметров хроматографической методики, которые могут быть внесены в нее для соответствия требованиям пригодности системы без принципиального изменения методик. Корректировка условий градиентного элюирования является более критичной, чем изократических условий, так как может вызвать сдвиг пиков на другую стадию градиента, и, таким образом, привести к некорректной идентификации пиков, наложению пиков или сдвигов, при которых элюирование интересующих соединений происходит после указанного времени регистрации хроматограммы. При внесении изменений, отличных от приведенных ниже, требуется проведение повторной валидации методики.
Проверка пригодности системы должна быть включена в методики для подтверждения получения разделения, требуемого для надлежащего проведения испытания. Тем не менее, поскольку неподвижные фазы описаны только в общем виде, и существует огромное количество доступных коммерческих фаз, отличающихся по хроматографическому поведению, некоторая корректировка условий хроматографирования может потребоваться для выполнения требований пригодности системы. В методиках с использованием обращенно-фазовой жидкостной хроматографии корректировки различных параметров не всегда приводят к удовлетворительному разделению. В этом случае может возникнуть необходимость замены одной колонки на другую, такого же типа, обеспечивающую желаемое хроматографическое поведение.
Если в фармакопейной статье не указано иное, допустимы следующие изменения параметров хроматографической системы.
Тонкослойная и бумажная хроматография
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
Пример 1: для вещества, содержание которого составляет 10 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 7 – 13 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению абсолютного содержания до 8 – 12 %. Допустимое изменение по относительному содержанию больше, чем допустимое изменение по абсолютному содержанию. Таким образом, допускается изменение содержания 7 – 13%.
Пример 2: для вещества, содержание которого составляет 5 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 3,5 – 6,5 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению его абсолютного содержания в подвижной фазе до 3 – 7 %. В этом примере допустимое изменение по абсолютному содержанию больше, чем допустимое изменение по относительному содержанию, и допустимым диапазоном содержания является 3 – 7 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Наносимый объём. При использовании пластинок с малым размером частиц сорбента (2 — 10 мкм) наносимый объём может быть изменен на 10 –20 % от заявленного объёма.
Жидкостная хроматография: изократическое элюирование
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Скорость потока подвижной фазы: ± 50 %, большая степень изменений допустима при одновременном изменении размеров колонки. При необходимости одновременного изменения размеров колонки и скорости потока сначала рассчитывается номинальная скорость потока для колонки, размеры которой отличаются от приведенной в нормативном документе, а затем допускается корректировка полученного значения скорости потока на ± 50 %.
Параметры колонки
- замена типа неподвижной фазы недопустима (например, недопустима замена фазы С18 на фазу С8);
- размер частиц: максимальное уменьшение размера частиц 50 %, увеличение не допускается.
Размеры колонки
Длина колонки: ± 70 %,
внутренний диаметр колонки: ± 25 %.
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

- F1 — скорость потока, указанная в нормативном документе, мл/мин;
- F2 — скорректированная скорость потока, мл/мин;
- l1 — длина колонки, указанная в нормативном документе, мм;
- l2 — длина используемой колонки, мм;
- d1 — внутренний диаметр колонки, указанный в нормативном документе, мм;
- d2 — внутренний диаметр используемой колонки, мм.
Температура: ± 10 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы (минимально определяемая концентрация) и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Жидкостная хроматография: градиентное элюирование
Изменение хроматографических условий для систем с градиентом требует большей осторожности, чем для изократических.
Состав подвижной фазы/профиль градиентного элюирования: незначительные изменения состава подвижной фазы и системы градиента являются приемлемыми при условии, что:
- выполнены требования пригодности системы;
- основной пик элюируется в пределах ± 15 % от обозначенного времени удерживания;
- элюирующая способность конечного состава подвижной фазы не менее предписанного состава.
Если при использовании градиентного элюирования не может быть достигнуто соответствие требованиям пригодности системы, рекомендуется оценить объем задержки градиента или сменить используемую колонку.
Объем задержки градиента. Конфигурация используемого оборудования может значительно изменить разрешение, указанные абсолютные и относительные времена удерживания. Это может произойти из-за отличия объема задержки градиента используемой системы от объема задержки градиента системы, с помощью которой проводилась валидация методики. В нормативном документе часто перед началом программы градиента включена изократическая стадия, чтобы можно было адаптировать систему к временным точкам градиента с учетом разницы объема задержки у системы, использованной для разработки методики, и у фактически использующейся системы. Решение о необходимости адаптации длительности изократической стадии для данного аналитического оборудования принимается пользователем. Если объем задержки, использованный при разработке методики, приведен в нормативном документе, то интервалы времени (t), указанные в таблице градиента, можно заменить адаптированными интервалами времени (tc), рассчитанными с помощью следующего уравнения:

D – объем задержки, мл;
D0 – объем задержки, использованный при разработке методики, мл;
F – скорость потока, мл/мин.
Изократическая стадия, введенная с этой целью, может быть исключена, если имеются данные валидации по применению методики без этой стадии.
pH среды водного компонента подвижной фазы. Изменения не допускаются.
Концентрация солей в буферном веществе подвижной фазы. Изменения не допускаются.
Скорость потока. Изменения допустимы при изменении размеров колонки.
Параметры колонки
- замена типа неподвижной фазы недопустима (например, недопустима замена С18 на С8);
- размер частиц: изменения не допускаются;
Размеры колонки
- длина колонки: ± 70 %;
- внутренний диаметр колонки: ± 25 %;
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

- F1 — скорость потока, указанная в нормативном документе, мл/мин;
- F2 — скорректированная скорость потока, мл/мин;
- l1 — длина колонки, указанная в нормативном документе, мм;
- l2 — длина используемой колонки, мм;
- d1— внутренний диаметр колонки, указанный в нормативном документе мм;
- d2 — внутренний диаметр используемой колонки, мм.
Температура. ± 5 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что детектирование (предел количественного определения) и сходимость отклика (RSD площадей или высот) для пика (пиков) определяемых соединений остаются удовлетворительными.
Газовая хроматография
Параметры колонки
- размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки);
- толщина плёнки: от минус 50 % до плюс 100 % (капиллярные колонки).
Размеры колонки
- Длина колонки: ± 70 %;
- Внутренний диаметр колонки: ± 50 %.
Скорость потока: ± 50 %.
Температура: ± 10 %.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Сверхкритическая флюидная хроматография
Состав подвижной фазы. Для набивных колонок количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше; для систем с капиллярной колонкой изменения не допускаются.
Длина волны детектора. Изменения не допускаются.
Параметры колонки
- размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки).
Размер колонки
- Длина колонки: ± 70 %;
- Внутренний диаметр колонки: ± 25 % (набивные колонки), ± 50 % (капиллярные колонки).
Скорость потока: ± 50 %.
Температура: ± 5 %, если температура указана в нормативном документе.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Хроматография. Лекция 1. Введение в хроматографию
Хроматография (определение IUPAC) – физический метод разделения, в котором компоненты смеси распределяются между двумя фазами, одна из которых (подвижная фаза) перемещается в определенном направлении относительно другой(неподвижной) фазы.
Принцип хроматографического разделения
Разделение происходит за счет разного скорости движения компонентов через слой сорбента. Эффект разделения основан на том, что компоненты хроматографируемой смеси проходят расстояние, на котором происходит разделение, с определенной для данного соединения задержкой.
Теоретически, скорость перемещения компонентов не должна зависеть от растворителя, природы веществ, их концентрации, но на практике, время от времени, принципы не применимы.
Хроматографический процесс – равновесный процесс, состоящий из элементарных актов сорбции и десорбции, растворения и элюирования, приводящих к новому состоянию равновесия.
Основная проблема хроматографического разделения: размывание пиков во времени. В градиентной хроматографии все пики узкие.
Классификация хроматографических методов
| Признак | Примеры |
| По агрегатному состоянию фаз | |
| По механизму межфазного распределения | |
| По способу проведения | |
| По способу перемещения сорбата | |
| По целям и задачам |
Классификация хроматографических методов по агрегатному состоянию фаз
Газовая хроматография
- Подвижная фаза – инертный газ (газ-носитель)
- Большое влияние оказывает температура
- Для хроматографирования летучих веществ и газов
- газо-твердофазная (газо-адсорбционная)
- газо-жидкостная
Жидкостная хроматография
- ПФ – жидкость
- Подходит для хроматографирования полярных веществ и макромолекул
- жидкостно-жидкостная
- жидкостно-твердофазная
- жидкостно-гелевая
Классификация хроматографических методов по механизму разделения (по характеру элементарного акта)
- Адсорбционная – основана на различной адсорбции веществ на поверхности сорбента
- Распределительная – основана на различной растворимости (абсорбции) веществ в ПФ и НФ
- Ионообменная – основана на различной способности к ионному обмену
- Хелатная – основана на различной способности к образованию хелатных комплексов
- Гель-фильтрационная (эксклюзионная, гель-проникающая) – основана на различной способности к проникновению в поры носителя. Вещества разделяются по размеру, первыми из колонки выходят вещества с большей молекулярной массой, так как они имеют больший размер и не задерживаются в порах.
- Хемихроматография – основана на различной реакционной способности. Скорость продвижения продукта реакции по НФ пропорциональна константе равновесия реакции.
- Аффинная – основана на различной биоспецифичности аналита и лиганда. Вещества, обладающие большим сродством к лигандам (молекулам, ковалентно связанными с НФ), задерживаются, в то время как остальные «смываются» подвижной фазой.
Классификация хроматографических методов по способу перемещения сорбатов вдоль слоя сорбента
Проявительная (элюентная) хроматография
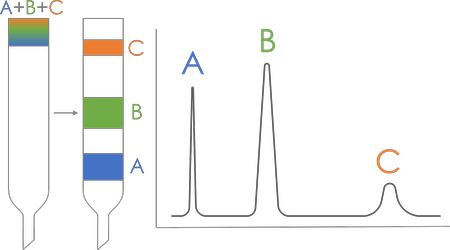
Хроматографируемая смесь делится в колонке на отдельные зоны, разделенные участками ПФ. Подходит для разделения многокомпонентной смеси.
Недостатки: требуется много растворителя
Фронтальная хроматография
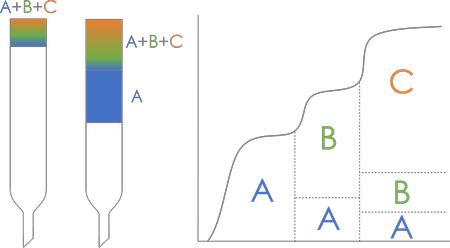
- эффективный метод
- требуется малое количество растворителя
Недостатки: только один компонент чистый
Применятся в установках по уменьшению жесткости воды, в респираторах, в промышленных фильтрах.
Вытеснительная хроматография
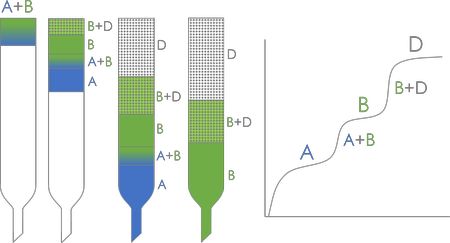
Используется не чистый растворитель, а вещество (вытеснитель) с высокой сорбционной способностью.
- высокая производительность
- требуется малое количество растворителя
- нет размывания зон
- скорость постоянна и равна скорости движения вытеснителя
Недостатки: большая продолжительность хроматографического процесса
Классификация хроматографических методов по способу проведения
- Колоночная
- Планарная
2.1 бумажная
2.2 тонкослойная
Классификация хроматографических методов по целям и задачам
- Аналитическая хроматография – получение информации (качественный и количественный анализ)
- Препаративная хроматография – разделение и очистка веществ
- Промышленная хроматография – автоматизированный контроль выбросов
Хроматографическая картина разделения
- Интегральная (практически не применяется)
- Дифференциальная
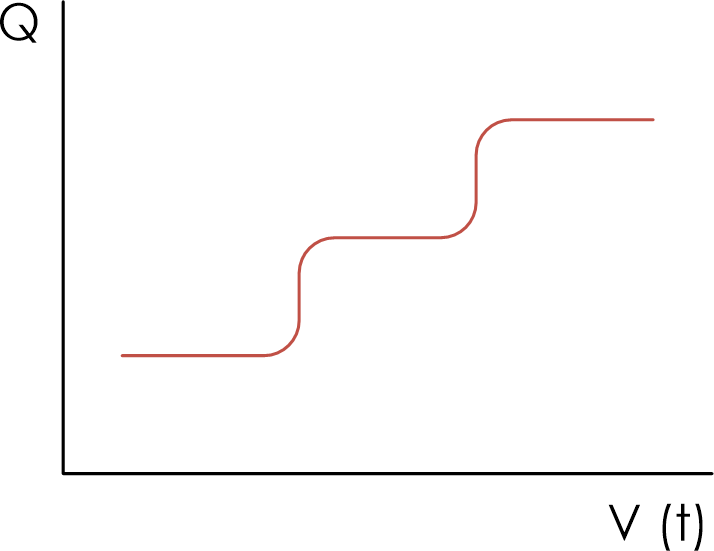
Интегральная выходная хроматографическая кривая,
Q=f(V) или Q=f(t).
Q – масса элюируемого вещества,
V – объем растворителя,
t – время хроматографирования.
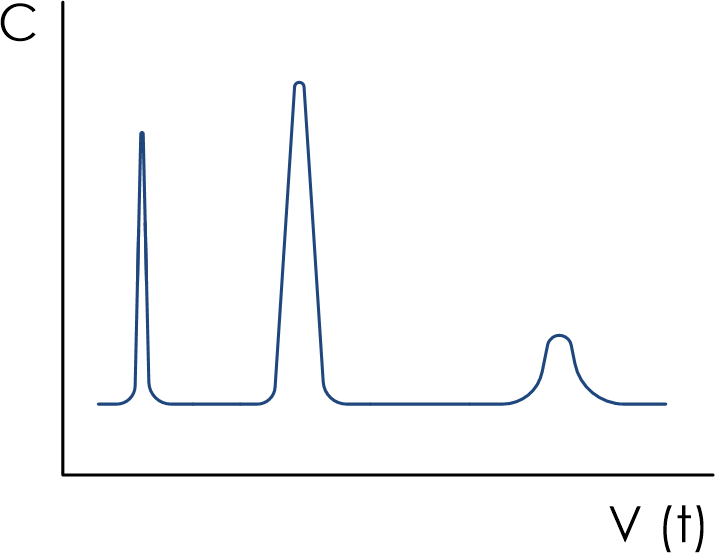
Дифференциальная выходная хроматографическая кривая,
C=f(V) или C=f(t).
C – концентрирования хроматографируемого
вещества.
Что такое хроматография? Типы и применения

Хроматография — это метод разделения и анализа смесей веществ, а также изучения физико-химических свойств веществ. Этот физический метод позволяет химикам внимательно наблюдать за органическими и неорганическими соединениями и выяснять, из чего они сделаны.
Слово «хроматография» означает «цветное письмо», но оно является неправильным, потому что оно часто не включает бумагу, чернила, цвет или письмо.
Метод был предложен в 1903 г. Михаилом Семеновичем Цветом — выдающимся русским исследователем. Первоначально свой метод M.С. Цвет назвал адсорбционным анализом (1903) и лишь через три года — хроматографическим методом (1906) 1 .

Михаил Семёнович Цвет (1872-1919)— русский ботаник-физиолог и биохимик растений, создатель хроматографического метода.
M.С. Цвет использовал хроматографичекий метод для разделения пигментов растений. Для разделения хлорофиллов Цвет наполнял стеклянную трубку (колонку) твердым адсорбентом (например, инулином) и наносил на верхний слой экстракт хлорофиллов в лигроине. Затем промывал колонку лигроином. Цвет писал так – «Из нижнего конца воронки вытекает сначала бесцветная, потом желтая жидкость (каротин), в то время как в поверхностных слоях инулинового столба возникает интенсивное зеленое кольцо, на нижнем крае которого быстро образуется желтая кайма.
При последующем пропускании через инулиновый столб чистого лигроина, оба кольца, зеленое и желтое, значительно расширяются и распространяются вниз до известного предела». «Как цветные лучи солнечного спектра различные компоненты из смеси пигментов были выделены и могли анализироваться дальше количественно и качественно». 2 Результат разделения, а именно последовательность различных цветовых зон Цвет назвал – хроматограммой. Для разделения пигментов Цвет использовал более ста различных адсорбентов, детально отработал технику разделения и предложил различные варианты аппаратов для своего метода (хроматографов).
Несколько десятилетий спустя открытия Цвета, ученые придумали новые виды хроматографии, различные сорбенты и хроматографическую технику.
Всемирно известно, что одно из открытий нового вида хроматографии, связано с нашей страной. В 1938 году в журнале «Фармация» вышла статья Н.А.Измайлова и М.С.Шрайбер «Капельно-хроматографический метод анализа и его применение в фармации» 3 , которая дала начало существования нового направления в хроматографии – тонкослойной хроматографии.
Последнее время появилось ряд сообщений авторитетных российских химиков о том, что практически параллельно с западными учеными первые работы в области аналитической газовой хроматографии выполнили в 1940-е г.г. советские исследователи М.М. Сенявин, Н.М. Тулькертауб, А.А. Жуховицкий и Д.А. Вяхирев. Это были работы по газо-адсорбционному разделению, выполненные задолго до широко известной публикации А. Джеймса и А. Мартина в 1952 г., от которой официально ведет отсчет история газовой хроматографии. 4
По экспертным оценкам, хроматография относится к 20 выдающимся открытиям прошедшего столетия, которые в наибольшей степени преобразовали науку, а через нее определили уровень развития техники и промышленности, цивилизации в целом. Хотя по образованию и роду занятий Цвет был ботаником, результаты его открытия столь значимы для всех естественных наук, что Федерация европейских химических обществ, например, приводит имя Цвета, наряду с четырьмя другими русскими именами — Ломоносова, Менделеева, Бутлерова и Семенова, — в числе ста выдающихся химиков прошлого. 5
Метод хроматографии основан на динамическом процессе распределения веществ между двумя фазами — неподвижной (твёрдая фаза или жидкость, связанная на инертном носителе) и подвижной (газовая или жидкая фаза, элюент). В зависимости от природы взаимодействия компонентов смеси с неподвижной и подвижной фазами и индивидуальных свойств, компоненты движутся с различной скоростью, что позволяет разделять их между собой.
Основные термины и понятия, относящиеся к хроматографии, а также области их применения были систематизированы и унифицированы специальной комиссией ИЮПАК. Согласно рекомендациям ИЮПАК, термин «хроматография» имеет три значения и используется для обозначения специального раздела химической науки, процесса, а также метода. 6
Существуют различные способы классификации хроматографических методов: по физическому состоянию подвижной фазы (газовая и жидкостная хроматографии), по технике выполнения хроматографического разделения (колоночная, плоскостная, хроматография в полях сил), по природе взаимодействия разделяемых компонентов с неподвижной фазой (адсорбционная, ионообменная, эксклюзионная и др.) и др.
Современная хроматография имеет много разновидностей, наиболее популярные их них, которые помогут вам получить более полное представление о процессе представлены ниже. Мы попытались объяснить их очень простым языком.
Основы хроматографии
По своей сути хроматография включает взаимодействие двух разных фаз. Химическое соединение в одном состоянии вещества (например, жидкость или газ) перемещается по поверхности другого вещества в другом состоянии вещества (например, твердое вещество или жидкость).
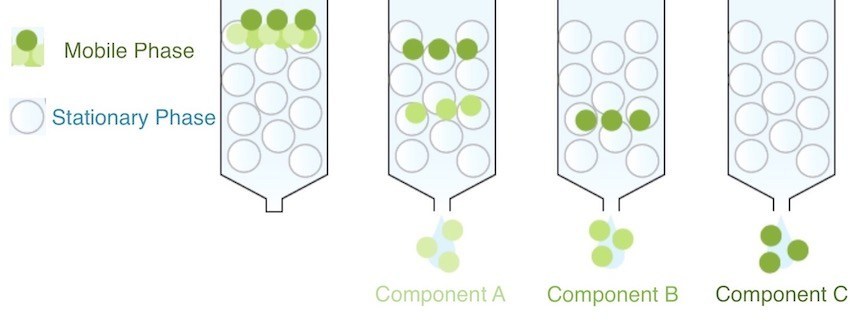
Движущееся соединение известно как подвижная фаза, в то время как устойчивое вещество (которое вообще не движется) называется стационарной фазой. Компоненты подвижной фазы отделяются, когда она движется по стационарной фазе. Затем химики могут анализировать отдельные компоненты один за другим.
4 разных типа хроматографии
Существует несколько видов хроматографии, каждый из которых имеет свой вид подвижной и стационарной фазы. Хотя основной принцип остается тем же самым, способ взаимодействия различных компонентов с подвижной фазой и стационарной фазой может варьироваться в зависимости от используемого хроматографического метода.
Ниже приведен список основных типов хроматографии, которые помогут вам получить более полное представление о процессе. Мы попытались объяснить их очень простым языком.
1. Бумажная хроматография

Бумажная хроматография является наиболее распространенным и простым аналитическим методом для разделения и обнаружения цветных компонентов, таких как пигменты. Хотя в современных лабораториях чаще используют тонкослойную хроматографию, он все еще является мощным учебным пособием.
В этом методе каплю образца смеси (например, чернил) помещают вблизи края фильтровальной бумаги, а затем бумагу подвешивают вертикально, при этом ее край погружают в растворитель (вода или спирт). Бумагу подвешивают таким образом, что пятно чернил не должно касаться растворителя и остается немного над ним.
Через некоторое время растворитель (подвижная фаза) начинает постепенно продвигаться вверх по бумаге (неподвижная фаза) посредством капиллярных сил. Поскольку растворитель движется вверх, он увлекает красители, присутствующие в чернилах, вместе с ним.
Когда он поднимается, мы видим разные цвета на фильтровальной бумаге. Эти цвета представляют различные красители, присутствующие в чернилах. Поскольку разные красители имеют разные уровни растворимости и движутся с разной скоростью, когда растворитель поднимается, мы видим полосы разного цвета на разной высоте.
Вот как бумажная хроматография используется для разделения разных компонентов чернил. В некоторых случаях смеси не содержат цветных компонентов, поэтому химики добавляют другие вещества для идентификации.
2. Тонкослойная хроматография

Тонкослойная хроматография очень похожа на бумажную хроматографию. Основное отличие состоит в том, что вместо куска бумаги у нас есть предметное стекло, покрытое слоем силикагеля (неподвижная фаза). В этом методе на нижний край предметного стекла с силикагелем наносятся капли раствора исследуемой смеси, лежащие на отрезке, параллельном нижнему краю и отстоящем от него на такое расстояние, чтобы капли не погружались в элюент.
Когда они подсохнут, предметное стекло нижним краем погружается в слой растворителя (элюент). Предметное стекло с неподвижной фазой удаляется из резервуара с растворителем, когда растворитель (подвижная фаза) достигает верхнего края стекла. Различные соединения в смеси перемещаются вверх по слою силикагеля с различной скоростью в виде пятен. Эти отделенные пятна затем визуализируются в ультрафиолетовом свете.
В некоторых случаях для визуализации пятен используются химические процессы: например, серная кислота обугливает большинство органических компонентов, оставляя темное пятно на предметном стекле. Это простая и быстрая техника для разделения смесей органических соединений. Она часто используется для определения пигментов, анализа состава красителей в волокнах и выявления инсектицидов или пестицидов в пищевых продуктах.
По сравнению с бумажной хроматографией, применение тонкослойной хроматографии приводит к лучшему разделению.
3. Газовая хроматография
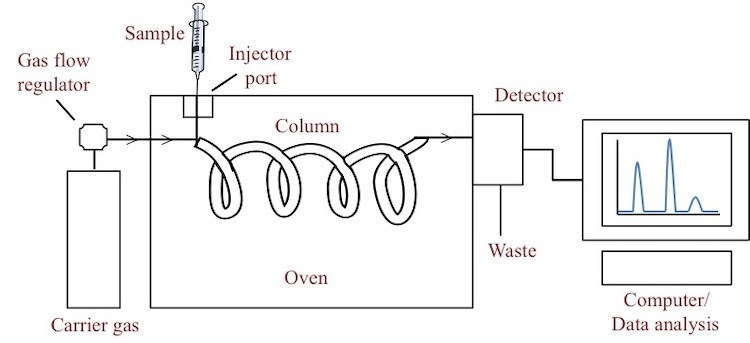
Газовая хроматография используется для разделения смесей летучих органических соединений. Прибор, выполняющий этот процесс, — газовый хроматограф — состоит из инжекционного порта, колонки с неподвижной фазой, детектора и системы регистрации данных. Смесь образцов (в газообразной форме) вводится через инжекционный порт.
Обычно количество пробы газа невелико, порядка микролитров. Подвижную фазу в газовой хроматографии называют газом-носителем. Поскольку мы не хотим, чтобы газ-носитель (подвижная фаза) реагировал с образцом, это должен быть инертный газ, такой как гелий, или нереакционноспособный газ, такой как азот. Колонка для газовой хроматографии (металлическая или стеклянная трубка) содержит неподвижную фазу тонкий слой жидкости или полимера на инертной твердой подложке.
Разделение компонентов в смеси происходит за счет разницы в их температурах кипения – соединения с низкой температурой кипения движутся быстрее компонентов с более высокой температурой кипения, а также за счет полярности и других специфических взаимодействий с подвижной фазой.
Это приводит к тому, что каждый компонент элюируется в разное время, также называемое временем удерживания компонента. Сравнивая времена удерживания разделенных компонентов с временами удерживания известных соединений, химики могут анализировать соединения в смеси.
4. Жидкостная хроматография
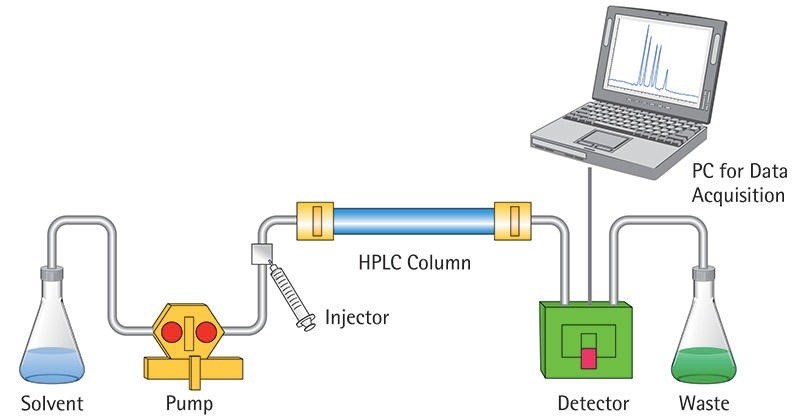
Жидкостная хроматография — это аналитический метод, используемый для разделения нелетучих соединений, находящихся в растворах в виде молекул или ионов. Его часто называют жидкостной хроматографией высокого давления, в которой подвижная фаза (растворитель) прокачивается через колонку с сорбентом под давлением.
Колонка обычно представляет собой металлическую или пластиковую трубку, заполненную крошечными частицами сорбента с определенным химическим составом поверхности. Поскольку каждое соединение в смеси по-разному реагирует с сорбентом (из-за различий в размерах, адсорбции и ионного обмена), они движутся в колонке с разными скоростями, что обеспечивает разделение их между собой. Выбор состава подвижной фазы зависит от свойств неподвижной фазы и анализируемых веществ.
Химики проводят серию тестов и отрабатывают методику разделения, чтобы найти оптимальный метод жидкостной хроматографии для смеси, который может обеспечить идеальное разделение пиков.
Вот быстрое сравнение четырех основных типов хроматографии —
| метод | Подвижная фаза | Неподвижная фаза (НФ) | Описание |
| Бумажная хроматография | Вода или органический растворитель | Бумага | Разделение за счет процессов распределения |
| Тонкослойная хроматография | Органический растворитель | Оксид алюминия или силикагель — на пластине | Разделение за счет процессов распределения и специфических взаимодействий с НФ |
| Газовая хроматография | Азот или гелий | Тонкий слой жидкости или полимера на инертной твердой подложке – в колонке | Разделение за счет разницы в температурах кипения и специфических взаимодействий с НФ |
| Жидкостная хроматография | Растворы | Сорбенты – в колонке | Разделение за счет специфических взаимодействий с НФ |
Применение
За научные исследования в области хроматографии или с применением хроматографического метода были присуждены несколько Нобелевских премий.
Более 60 процентов химических исследований во всем мире проводится с помощью различных видов хроматографии. Современные хроматографы способны разделить и идентифицировать несколько сотен соединений за один анализ. Некоторые хроматографические детекторы могут определять количество вещества в масштабе ppb.
Благодаря этим преимуществам, хроматография в настоящее время широко используется в
- Криминалистика: анализ образцов, полученных с мест преступления
- Мониторинг загрязнений: для обнаружения небольших концентраций опасных загрязнителей в воздухе и воде.
- Медицинская сфера: в процессе производства и контроле качества биологических и фармацевтических продуктов.
- Пищевая промышленность: обнаружение порчи в пищевых продуктах, определение качества продуктов питания, а также контроле пищевых добавок.
- Юридические действия: определить наличие алкоголя в крови и кокаина в моче.
- Радиохимия: для характеристики радиоактивно меченных соединений и определения радиохимической чистоты.
Помимо этого, хроматография также используется для расшифровки ДНК и в биоинформатике, клинической диагностике заболеваний и расстройств, а также в различных исследовательских целях.
1 Е.М. Сенченкова. Михаил Семенович Цвет. Москва: Издательство «Наука», 1973
2 М.С. Цвет «Хроматографический адсорбционный анализ. Избранные работы. Под ред. А.А. Рихтера и Т.А. Красносельской. Изд-во АН СССР. 1946
3 Измайлов Н.А., Шрайбер М.С.. Капельно-хроматографический метод анализа и его применение в фармации. Фармация. 1938, №3.с.1-7
4 Р.Х. Хамизов, В.Ф. Селеменев. Кто открыл газовую хроматографию? // Сорбционные и хроматографические процессы. 2018. Т. 18. № 2. С 128-130
5 «Сто лет хроматографии» В. А. Даванков, Я. И. Яшин // Вестник РАН, 2003, том 73, № 7, с. 637-646
6 Nomenclature for Chromatography // Pure and Appl. Chem. 1993.Т. 65, № 4. С. 819—872
http://studentoriy.ru/xromatografiya-lekciya-1-vvedenie-v-xromatografiyu/
http://new-science.ru/chto-takoe-hromatografiya-tipy-i-primeneniya/