Термодинамика процессов диссоциации
В практике пирометаллургии цветных металлов исходным сырьем являются: руда, концентраты, промежуточные продукты металлургического производства, в которых ценные компоненты находятся в виде химических соединений: оксидов, сульфидов, галогенидов, карбонатов и др. Металлы могут быть извлечены из этих соединений за счет простейшей химической реакции (термической диссоциации соединений):
где Ме — металл; Х — анион.
Основоположником теории термической диссоциации соединений справедливо считают академика А.А. Байкова, который еще в 20-е годы изложил основные принципы этого процесса. Для анализа процесса А.А. Байков предложил воспользоваться термодинамическим методом. В условиях равновесия в изолированной системе Ме-X при давлении, равном 10 5 Па (в пирометаллургии вакуумные процессы еще не нашли широкого применения) при любой температуре в газовой фазе должно находиться равновесное количество катионов Ме, анионов Х и паров самого соединения: Кр = РMеРХ/РМеХ
Реакция диссоциации требует затраты тепла и поэтому в соответствии с принципом Ле-Шателье при повышении температуры равновесие реакции должно смещаться в сторону образования продуктов реакции.
Многие металлы имеют несколько форм валентности. При диссоциации их соединений могут образовываться промежуточные соединения низшей валентности. А.А. Байковым в свое время был сформулирован так называемый принцип последовательности превращения. Рассмотрим эту схему на примере диссоциации какого-либо оксида металла:
Здесь b1, b2, n, m, q, р — стехиометрические коэффициенты реакции.
Согласно принципу А.А. Байкова, при равновесном разложении оксида отрыв кислорода от соединения происходит последовательно, скачками, проходя через все те химические соединения, которые могут существовать в системе. Например, диссоциация гематита до железа в равновесных условиях при температуре >570 °С (см. диаграмму) должна проходить по схеме Fе2O3-Fе3O4-Fе1-xO-Fе.
На основании предыдущего изложения мы уже убедились в том, что любое соединение имеет большую или меньшую область гомогенности. Это вносит свои особенности в теорию диссоциации соединений, что и было в свое время отражено в классических трудах А.А. Байкова.
В настоящей лекции рассмотрим термодинамику диссоциации соединений для условий, когда внешнее давление принято равным 10 5 Па. Это — реальное состояние большинства пирометаллургических агрегатов в настоящее время.
Диссоциация соединений переменного состава при постоянной температуре
Диссоциация соединения связана с переходом летучего компонента в газовую фазу. Прежде всего, необходимо выяснить, какая же реакция при этом протекает? Вопрос этот не является таким простым, как это может показаться с первого взгляда. Возьмем сначала группу соединений с одним летучим компонентом (оксиды, сульфиды, хлориды нелетучих металлов). Для этой группы веществ нужно раздельно рассматривать диссоциацию соединения, лежащего на металлическом крае области гомогенности и внутри области гомогенности. допустим, что был взят вюстит, лежащий на железном крае области гомогенности (см. диаграмму Fe-O). Диссоциация этого соединения, т.е. переход кислорода в газовую фазу при постоянной температуре (1200 °С) приведет к тому, что фигуративная точка, определяющая состояние системы, будет перемещаться в зону устойчивости твердого раствора кислорода в γ-Fe по изотерме t.
Равновесное давление кислорода над системой остается постоянным до тех пор, пока полностью не исчезнет одна из конденсированных фаз. Если давление кислорода в реакционном сосуде будет меньше, чем равновесное давление над вюститом этого состава, то исчезнет фаза вюстита, произойдет его диссоциация. Процесс этот может быть выражен реакцией:
Если давление кислорода в реакционном сосуде будет больше равновесного, то реакция пройдет в правую сторону до исчезновения металлической фазы. Важно еще раз подчеркнуть, что при диссоциации вюстита образуется не чистое железо, а железо, насыщенное кислородом. Константа равновесия реакции равна: Кр, = рO2 1/2 рMe /рFe1-xО;
Допустим, что имеется вюстит, состав которого отвечал кислородному краю области гомогенности (диаграмма, точка b), При этом диссоциация соединения, отвечающего точке b, т.е. переход кислорода в газовую фазу и уменьшение его содержания в вюстите вовсе не сопровождается появлением металлической фазы. Фигуративная точка, определяющая состав системы, лишь перемещается внутрь области гомогенности (точка d). При этом уменьшение содержания кислорода приводит к тому, что возрастает отношение железа к кислороду, т.е. уменьшается количество вакансий железа. Величина x в формуле Fe1-xO уменьшается, но вакансии железа двузарядны и исчезновение каждой вакансии сопровождается исчезновением двух дырок.
Увеличение (снижение) концентрации точечных дефектов обуславливает резкое возрастание (уменьшение) равновесного давления летучего компонента. Даже для соединений с узкой областью гомогенности равновесное давление аниона веществ, расположенных на ее краях, отличается на несколько порядков.
Диссоциация соединений с образованием конденсированных фаз постоянного состава
Остановимся более подробно на том случае, когда оксид и металл образуют две фазы постоянного состава, находящиеся в твердом состоянии. В рассматриваемой системе число компонентов равно двум, число фаз — трем. Тогда число степеней свободы системы равно единице (в соответствии с правилом фаз): С = К – Ф +2 = 2 — З +2 = 1. Это значит, что для полного описания состояния такой равновесной системы достаточно одного параметра. В термодинамике состояние системы, как правило, описывается параметрами Р и Т, но один из них является зависимым. За независимый параметр, как правило, принимают температуру. Тогда общее давление в рассматриваемой системе при заданных температуре и составе фаз будет равным:
Качественно зависимость равновесного давления от температуры можно охарактеризовать с помощью правила Ле-Шателье. Реакция диссоциации соединений является эндотермической. Подвод тепла извне приводит к разложению дополнительного количества вещества до тех пор, пока нарастающее в системе противодействие, выражающееся в конкретном случае в повышении рO2, не приведет к установлению нового равновесного состояния. Напротив, если в системе повышать давление, равновесие реакции диссоциации сдвинется в левую сторону.
Проанализируем вид функции, исходя из следующих соображений. В общем виде константа равновесия реакции диссоциации может быть записана в виде:
Допустим, что при условиях протекания процесса aMe и aMeO — величины постоянные. Тогда К = PO2.
Для определения зависимости константы равновесия реакции диссоциации (а значит и упругости диссоциации) от температуры можно воспользоваться уравнением изобары:
 |
где:
Для практических нужд нередко можно воспользоваться упрощенным уравнением, считая, что ΔH и Ср в исследуемом относительно узком интервале температур мало зависят от температуры. Тогда lnKp = − ΔH/RT + const.
Графически уравнение состояния рассматриваемой системы будет изображаться логарифмической кривой (рисунок 4.1). Поле диаграммы pO2 – T разделено на две области. Кривая на рисунке представляет собой геометрическое место точек, характеризующих равновесное давление в системе с чистыми металлом и оксидом.
Если в исходном состоянии в рассматриваемой изолированной системе давление кислорода было больше равновесного давления рO2‘ >pО2 при температуре Т, то система находилась в неравновесном состоянии (рO2’ — исходное давление кислорода в системе; рO2 — равновесное давление). Стремление системы к снижению свободной энергии и к переходу в равновесное состояние пойдет за счет взаимодействия кислорода с металлом. Изменение изобарного потенциала реакции диссоциации в случае, если внешнее давление не равнялось единице, описывается уравнением:
 Так как рO2‘> рO2, а RТ> 0, то ΔG > 0. Согласно термодинамике, реакция диссоциации в данном случае не будет иметь места. Наоборот, произойдет обратная реакция — окисление металла кислородом. Переход системы в равновесное состояние в изотермических условиях при отводе выделяющегося вследствие окисления металла тепла можно описать на графике прямой аа’ (изотермические условия). Если процесс осуществляется при сохранении постоянного давления (изобарические условия), то переход системы в равновесное состояние пройдет по линии аa”. Если же ни одно из этих условий не будет соблюдаться достаточно строго, то переход системы в равновесное состояние определяется какой-то траекторией аa’”.
Так как рO2‘> рO2, а RТ> 0, то ΔG > 0. Согласно термодинамике, реакция диссоциации в данном случае не будет иметь места. Наоборот, произойдет обратная реакция — окисление металла кислородом. Переход системы в равновесное состояние в изотермических условиях при отводе выделяющегося вследствие окисления металла тепла можно описать на графике прямой аа’ (изотермические условия). Если процесс осуществляется при сохранении постоянного давления (изобарические условия), то переход системы в равновесное состояние пройдет по линии аa”. Если же ни одно из этих условий не будет соблюдаться достаточно строго, то переход системы в равновесное состояние определяется какой-то траекторией аa’”.
Если парциальное давление кислорода в изолированной системе при температуре Т2 ниже равновесного (рO2’’ . 10 5 Па) наименее устойчивым оксидом будет Аg2О. Действительно, процесс диссоциации оксида, будет иметь место в том случае, когда в системе будет меньше равновесного значения упругости диссоциации. Этот момент наступает для Аg2О при температуре 450 °С, для СuО при 1300 °С. На рисунке 4.2 представлены температурные зависимости упругости диссоциации ряда оксидов и сульфидов, позволяющие оценить вероятность нахождения металлов при определенной температуре и парциальном давлении кислорода, серы в свободном состоянии или в виде соединения. У благородных металлов упругость диссоциации их соединений уже при низких температурах составляет значительную величину. Поэтому такие металлы встречаются в земной коре в свободном состоянии в виде самородных, что нельзя сказать, например, о самородном железе. Зависимость упругости диссоциации FeО от температуры выражается уравнением:
Диссоциация монооксида железа начнется, когда рO2’≤ рO2. В воздушной атмосфере рO2 = 0,21 . 10 5 Па. Для равновесных условий протекание реакции диссоциации 2ЕеО↔ 2Fe + O2 монооксида железа на металл и кислород начнется при температуре 3760 К. При более низких температурах идет обратный процесс, результатом которого будет образование прочных оксидов железа.
 |
Несомненно, более объективной характеристикой сродства металлов к сере, кислороду или хлору служит убыль изобарного потенциала реакции образования соединения из элементов, отнесенная к стандартным условиям. Изменение изобарного потенциала реакнии окисления может быть описано следующим уравнением: ΔG = RT (lnKp’-lnKp) [Kp’ — величина, характеризующая исходное состояние системы (формально записывается так же, как константа равновесия реакции, однако величины, составляющие ее, относятся к неравновесному исходному состоянию)].
Чтобы было удобно сравнивать между собой величины убыли изобарного потенциала образования различных соединений из элементов, за стандартное исходное состояние системы принимают давление 10 5 Па паров серы или кислорода (анионов) в системе. Константа равновесия реакции окисления (величина обратная диссоциации) металлов для случая нахождения металла и оксида в конденсированных фазах при постоянстве их состава запишется как К=1/PO2. Тогда, нормальное сродство металла к кислороду определяется из выражения: ΔG = RT (ln1 — ln(pO2 -1 )) = — RT ln(pO2 -1 ) (рO2 — упругость диссоциации оксида).
Величину изменения термодинамического потенциала реакции можно отнести к различному количеству реагирующих веществ. При выражении значения ΔGобразования оксидов и сульфидов из элементов как меры химического средства численное значение убыли термодинамического потенциала всегда относят на 1 молы кислорода или серы.
На рисунке 4.3 приведены температурные зависимости убыли стандартного термодинамического потенциала образования соединений, отвечающих по составу краю области гомогенности. Чем более отрицательные значения имеют величины тем больше сродства у этих металлов к кислороду и к сере, меньше упругость диссоциации соединений, более прочным будет оксид или сульфид. Наоборот, чем положительнее значения убыли термодинамического потенциала образования соединения для какого-либо металла, тем меньше его химическое сродство к кислороду и сере, тем менее прочны оксиды и сульфиды этого металла. Наибольшее сродство к кислороду имеют такие элементы, как кремний, ванадий, алюминий, цирконий, барий, кальций. Наименьшее — серебро, ртуть, медь, свинец.
Наибольшим сродством к сере при высоких температурах отличаются медь, цинк и кальций
 | 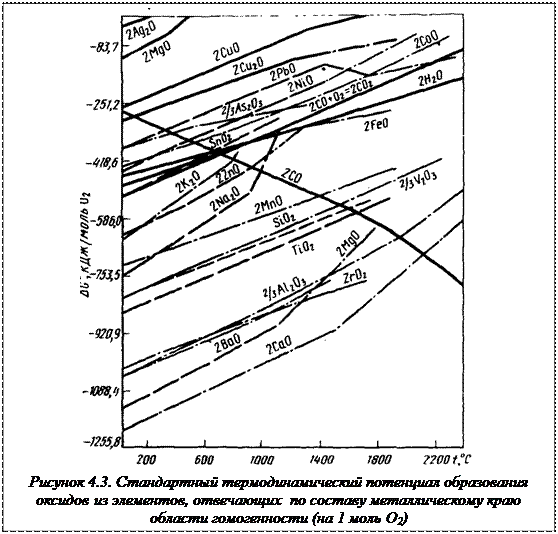 |
Сродство к кислороду у металлов существенно выше, чем к сере. По этой причине при протекании обменной реакции типа Ме’О + Ме”S = Ме’S + Ме”О решающее значение оказывает разность в величинах сродства металлов к кислороду. Например, кальций концентрируется в окисленной фазе, несмотря на его высокое сродство к сере.
При диссоциации высоких оксидов, сульфидов или хлоридов происходит первоначально их разложение на соединения более низкой валентности.
Диссоциация оксидов и сульфидов, образующие растворы
Если соединение и металл образуют друг с другом или с посторонним инертным веществом растворы, то вместо двух конденсированных фаз образуется одна. Число степеней свободы у такой системы равно двум.
Упругость диссоциации соединений будет функцией уже не только температуры, но и состава
Геометрическое выражение этой зависимости — поверхность. Рассмотрим на примере оксида двухвалентного металла функциональную зависимость упругости диссоциации. В системе из двух несмешивающихся фаз: металлического раствора и раствора диссоциирующего соединения. Уравнение реакции в этом случае имеет следующий вид:
где [Ме] и (МеО) — концентрации соответственно металла и оксида в растворе.
В качестве исходных предпосылок примем, что растворители для металла и оксида инертны по отношению к кислороду. Пусть растворимость металла и оксида будет ограниченной. Предельные концентрации в насыщенных растворах обозначим как [Ме]н.р. и (МеО)н.р.. Константу равновесия реакции можно записать следующим образом:
где: РМе и РМеО — равновесные парциальные давления соответственно пара металла и оксида в системе при температуре Т.
Для рассматриваемого случая упругость диссоциации равна:
Согласно закону Геири равновесное давление пара вещества над раствором пропорционально молярной доле его в растворе.
Давление пара вещества над насыщенным раствором равно давлению пара чистого вещества (РMe 0 , рMeO 0 ). Тогда можно записать:
Подставляя значения упругости диссоциации оксида для системы с раствором, получим:

Выражение определяет упругость диссоциции оксидов в том случае, когда металл и оксид растворяются в каких-то растворителях.
Так как  , т.е. константа диссоциации веществ K0, не образующих растворы, то:
, т.е. константа диссоциации веществ K0, не образующих растворы, то:
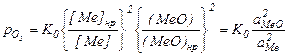
Где: аMеO и аMe активности оксида и металла в растворах Если металл остается в конденсированной фазе, а в раcтворе находится только оксид, выражение упрощается. Так как [Ме]н.р. /[Ме] = 1, то:
Из выражения следует, чторавновесная упругость диссоциации оксида находится в зависимости от его концентрации (активности) в растворе. Чем меньше концентрация оксида в растворе, тем меньше упругость его диссоциации, тем прочнее оксид и тем труднее его восстановить до металла. При повышении концентрации равновесное давление возрастает лишь до величины, соответствующей рO2 для конденсированного нерастворенного оксида.
Рассмотренная зависимость имеет большое значение в практике пирометаллургии. Например, при высокотемпературном восстановлении оксидов цветных металлов процесс восстановления будет протекать тем легче, чем выше концентрация оксида в расплаве. По этой причине при восстановлении трудно восстановимых оксидов, т.е. имеющих большое сродство к кислороду, стремятся иметь высокую концентрацию их в расплаве Не менее часто встречается задача выделения небольших количеств ценных компонентов из оксидного и силикатного расплавов. Очевидно, при снижении концентрации оксидов в расплаве упругость их диссоциации тоже будет снижаться, что потребует и более жестких условий восстановления. Если в растворе находится образующийся при диссоциации металл, а оксид представлен индивидуальным веществом, то уравнение преобразуется следующим образом: рO2 = К0 ([Ме]н.р. / [Ме]) 2 .
Из этого выражения следует, что упругость диссоциации оксида, когда образующийся металл растворяется в инертном растворителе, всегда больше или равна упругости диссоциации оксида, когда металл находится в элементарной форме.
Выведение образующегося металла в раствор обеспечивает более полное восстановление оксидов, протекающее с меньшими затруднениями. В практике пирометаллургии с образованием металлических растворов приходится встречаться при термическом получении силумина, оловянной электроплавке и т.п.
Теоретические основы.
Читайте также:
|
| твердое тело | С0 V1 | растворитель С V2 |
B ________δ_________ E
Рис. 1. Растворение твердого вещества в жидком растворителе (С0 – концентрация вещества в диффузном слое; С – концентрация вещества в растворителе; δ – толщина диффузного слоя; V1 – скорость растворения твердого тела в диффузном слое; V2 – скорость диффузии растворенного вещества в растворителе).
Процесс выравнивания концентраций по всему объему идет очень медленно. Перемешивание ускоряет этот процесс.
При перемешивании выравниваются концентрации в большей части объема, но у самой поверхности раздела всегда остается небольшой неперемешивающийся слой (δ), называемый диффузным.
Изменение концентрации в приповерхностном слое показано на рис.2.

Рис.2 Изменение концентрации в приповерхностном слое (АВЕD).
Рассматривая скорость процесса растворения твердого тела в жидкости, можно принять, что в части, непосредственно прилегающей к твердому телу (АВ рис.1,2), слой в большей степени приближается к состоянию равновесия с твердым телом, и концентрация растворенного вещества в нем приближается к концентрации насыщенного раствора (Со). В части слоя, прилегающей к внутреннему объему жидкости (ДЕ рис1,2), концентрация растворенного вещества приближается к концентрации С в остальном объеме жидкости.
Скорость диффузии (V1, рис. 1) будет тем больше, чем больше различия в концентрации (Со – С) диффундирующего вещества.
Количественно скорость растворения может быть выражена уравнением:
n — число молей диффундирующего вещества;
τ – время диффузии; D – коэффициент диффузии;
S – величина поверхности соприкосновения фаз;
δ – толщина диффузного слоя;
Со – концентрация растворенного вещества в насыщенном слое (АВ, рис 1,2):
С – концентрация растворенного вещества в объеме раствора.
Изменение числа молей dn можно заменить соотношением
где V — объем раствора; dC – изменение концентрации в объеме раствора.
Подставив уравнение (2) в (1), получим:
Обозначим постоянные для данных условий величины через константу (К):
тогда уравнение (3) примет вид:
Полученное уравнение идентично кинетическому уравнению реакции первого порядка:
Полученное уравнение представляет собой уравнение прямой линии типа у = εх, (10) выходящей из начала координат.
Если построить график в координатах ln Со/Со-С=f(τ) (рис.3), то получим прямую линию ОВ, выходящую из начала координат. Тангенс угла φ наклона этой линии (ОВ) к оси абсцисс равен константе К. Построив любой прямоугольный треугольник с гипотенузой на линии ОВ, можно определить константу скорости процесса растворения твердого тела:

К = tgφ = | ВС| / |АС| (11)
Величины катетов (ВС и Ас) берут, учитывая масштаб графика.

Рис. 3 Зависимость ln Cо/Со-С
2.3. Определение порядка реакции.
Под порядком понимают сумму показателей степеней при концентрациях в уравнении скорости реакции (6). Порядок реакции проще всего определяется методом подбора уравнений.
Метод подбора уравнений основан на подстановке концентрации регулирующего (растворяющегося) вещества для каждого момента времени о начала реакции в кинетические уравнения первого, второго порядков. Порядок реакции соответствует уравнению, для которого рассчитанная константа в различные моменты времени остается постоянной.
Графический метод определения порядка реакции заключается в определении такой функции, согласно которой зависимость от времени выражается прямой линией. Для реакции первого порядка такой зависимостью является ln Cо/(Со-С) = f(C) или
ln Cо/С=f(C). Для реакции второго порядка (Со-С)/(Со-С)= f(C) или 1/С = f(C).
2.4. Влияние температуры на скорость реакции. Энергия активации.
Для всякого процесса существует промежуточная стадия перехода нормальных молекул в активные, между которыми устанавливается равновесие.
Энергия активации (Е) – это то избыточное количество энергии (по сравнению со средней величиной), которой должна обладать молекула, чтобы быть способной к данному взаимодействию. С увеличением температуры количество активных молекул увеличивается.
Влияние температуры на процесс растворения твердого тела можно выяснить, определяя константы скорости при различных температурах. Скорость почти всех процессов возрастает с повышением температуры. Отношение констант скоростей при двух температурах, отличающихся на 10, называется температурным коэффициентом γ:
где Кt+10 — константа скорости при температуре t+10 o ,
Kt — константа скорости процесса при температуре.
где К – константа скорости процесса
Т – температура в градусах Кельвина
Е – энергия активации (Дж/моль)
R – универсальная газовая постоянная (8,31 Дж/моль. град.).
При проведении процесса при двух температурах (T1 и Т2) можно дифференциальную форму уравнения Аррениуса представить в интегральном виде:
где K1 и К2 — константы скорости при температурах T1 и T2
Дата добавления: 2015-08-05 ; просмотров: 9 ; Нарушение авторских прав
Зависимость скорости реакции от температуры
Для характеристики влияния температуры на скорость реакции для небольшого температурного интервала обычно используют температурный коэффициент (γ), представляющий собой отношение скоростей реакции или констант скорости реакции при двух температурах, отличающихся на 10º.
Правило Вант-Гоффа установлено экспериментально. Для гомогенных реакций:
γ = WТ+10 / WТ =2¸4или γ = kТ+10 / kТ =2¸4
При повышении температуры на 10º константа скорости химической реакции увеличивается в 2-4 раза.
Химическая реакция, протекающая в пределах одной фазы, называется гомогенной химической реакцией (пример, любая реакция в растворе).
Химическая реакция, протекающая на границе раздела фаз, называется гетерогенной химической реакцией (пример, любая реакция, идущая на поверхности твердого катализатора — гетерогенная каталитическая реакция). Для гетерогенной реакции γ = 1,1¸1,3 (лимитирующей стадией является процесс диффузии).
Правило Вант-Гоффа является приближенным, так как температурный коэффициент принимают за постоянную величину. В действительности температурный коэффициент изменяется с температурой, при повышении температуры γ→1.
Более точную зависимость влияния температуры на скорость химического процесса дает уравнение Аррениуса. Это основное уравнение химической кинетики:
dlnk/dT = E /RT 2 — уравнение Аррениуса в дифференциальной. форме или
dlnk = E dT /RT 2 .
Проинтегрируем: lnk = — E/RT + const.
Пусть const = Н, тогда lnk = Н — E/ RT,
lnk = (Н — E/ RT)´lne = lne ( Н — E/ RT) ,
k = e H e ( — E/ RT) = A e ( — E/ RT) .
Уравнение Аррениуса в интегральной форме.
k= A e — E/ RT ,
где А — предэкспоненциальный множитель, который постоянен для данной реакции и данных условий (величина, практически не зависящая от температуры); е — основание натурального логарифма; Е — энергия активации.
Интегрирование уравнения Аррениуса в пределах температур Т1 и Т2 дает соотношение:
Перейдем к десятичному логарифму:
Зная энергию активации и хотя бы одно значение контакта скорости, можно рассчитать константу скорости для любой другой температуры.
5.6. Теории химической кинетики. Теория активных соударений
Задачи теорий химической кинетики: 1) выяснение механизма химических реакций; 2) выяснение влияния различных факторов на скорость реакции (температура, концентрация, давление, катализатор и др.); 3) теоретический расчет константы скорости.
Для теоретического расчета констант скоростей используют две теории: 1) теория активных соударений или теория бимолекулярных реакций Аррениуса (1889 г.); 2) метод активного комплекса или теория переходного состояния (Эйринг и Поляни, 1935 г). Эта теория сохранила значение и по настоящее время.
Теория активных соударений была предложена Аррениусом для бимолекулярных газовых реакций. Согласно кинетическим представлениям для начала реакции необходимо столкновение реагирующих молекул. Чем выше концентрация, тем больше число столкновений и тем больше скорость реакции: W = kc1´c2. Однако опыт показывает, что реакция всегда протекает со значительно меньшей скоростью, т.е. столкновение молекул является необходимым условием, но не достаточным: не каждое столкновение приводит к реакции. Например, разложение йодистого водорода:
Из 10 17 столкновений лишь одно приводит к образованию продуктов реакции. Если скорость реакции зависит лишь от числа соударений, то нельзя объяснить влияние температуры на скорость реакции, так как при повышении температуры на 10º число столкновений увеличивается на 1-2 %, а скорость реакции возрастает в 2-4 раза.
По Аррениусу каждая химическая реакция протекает в две стадии:
нормальные молекулы « активные молекулы ® продукты реакции.
Аррениус принимал за активные молекулы особую химическую модификацию нормальных молекул. Аррениус не объяснил истинную природу активных молекул, физический смысл предэкспоненциального множителя “А” в уравнении: k = Ae — E/ RT .
Дальнейшее развитие теории активных соударений было сделано Д.В. Алексеевым (1915-24 г.г.). Он предложил активными считать молекулы, обладающие повышенной энергией по сравнению со средней энергией молекул, и для объяснения природы активных молекул использовать законы Максвелла (закон распределения молекул по скоростям) и Больцмана (закон распределения молекул по энергиям). Закон Максвелла дает возможность определить, какая доля общего числа молекул в данных условиях обладает скоростью, точно отвечающей средней скорости, и какая отличается от нее на ту или другую заданную величину. В математической форме этот закон выражается сложным соотношением. Мы ограничимся здесь разбором только графического выражения этой зависимости (рис. 5.3.):
dN/Ndυ — доля молекул dN, обладающих скоростями в пределах между “W” и
“W + dW” от общего числа молекул N.

Рис. 5.3. Распределение молекул по скорости.
Кривые имеют четко выраженный максимум и сильнее растянуты в сторону больших скоростей. Скорость “W” называется наиболее вероятной, она не совпадает со средними скоростями. По Алексееву активные молекулы — это молекулы, располагающиеся в хвосте Максвелловской кривой, т.е. обладающие избыточной скоростью. Кривая распределения скоростей данного газа зависит только от температуры. При повышении температуры скорость молекул возрастает. Максвелловская кривая сдвигается вправо, одновременно уменьшается максимум, но доля молекул, обладающих избыточной скоростью резко возрастает (площадь, ограниченная кривой и осью “W”). Максвелловская кривая количественно описывается уравнением Больцмана. Для частного случая, когда энергия молекул зависит только от двух степеней свободы (движение в плоскости) уравнение имеет вид:
где NE — число молекул с энергией не меньше “Е”, N0 — обще число молекул.
Такая же зависимость для числа двойных столкновений:
Правый член (e — E/ RT ) — фактор Больцмана (фактор активации):
где ZE — число активных соударений, Z0 — общее число соударений.
С повышением температуры число активных соударений резко увеличивается. Основное положение теории активных соударений: реагируют лишь те молекулы, энергия которых в момент столкновения не ниже некоторого предела “Е”, называемого энергией активации.
Таким образом, энергией активации называется та минимальная избыточная энергия по сравнению со средней величиной, которой должны обладать молекулы, чтобы их столкновение привело бы к реакции.
Рассмотрим ход реакции в зависимости от энергетического состояния молекул (рис. 5.4.):
Е1 — энергия исходных веществ, Е2 — энергия продуктов реакции, Е3 — наименьший запас энергии, которым должны обладать молекулы, чтобы их столкновения могли привести к химическому взаимодействию.

Рис. 5.4. Изменение энергии реакционной системы.
Данная реакция экзотермическая, т.е. общий запас энергии продуктов реакции меньше, чем исходных веществ: ΔН = Е2 — Е1 — E/ RT .
Для многих реакций υ -E/RT ,
где p — стерический коэффициент (фактор вероятности). Значение “p” меняется в пределах от 1 до 10 -9 и теоретически не может быть рассчитано. Все это следствие несовершенства и формального характера теории активных соударений, которая рассматривает молекулы реагирующих веществ как жесткие шарики.
Например, реакция этерификации замещенных бензойных кислот в присутствии концентрированной минеральной кислоты:
R-Ph-СOOH + C2H5OH ® сложный эфир.
При R=Н реакция протекает легко. Если в орто-положение к карбоксильной группе ввести заместитель (R = NO2), то реакция вообще не пойдет. Скорость реакции понижается в ряду R = F, Br, Cl, J.
2. Для сильно экзотермических реакций выделяющаяся энергия может привести к распаду продуктов реакции на исходные вещества.
3. При слишком кратковременных соударениях сложных молекул энергия не успевает перераспределиться должным образом и привести к разрыву нужных связей.
http://lektsii.com/2-119730.html
http://mylektsii.su/2-71873.html