«Термодинамика химического равновесия Лекция 3 Фундаментальные уравнения для закрытых систем (дифференциалы термодинамических . »
Фундаментальные уравнения для
закрытых систем (дифференциалы
• Для закрытой системы постоянного состава, в которой может
совершаться только работа расширения:
• Q = dU + pdV; Q = TdS; TdS = dU + pdV; dU = TdS – pdV
• Это уравнение называют фундаментальным уравнением для
закрытой системы постоянного состава, которая может
совершать только работу расширения.
• Частные производные внутренней энергии:
а) V=const; dV=0; dU=TdS; (U/S)V=T
б) S=const; dS=0; dU=-pdV; (U/V)S= -p
• Полный дифференциал функции U:
dU = (U/S)V·dS + (U/V)S·dV
• Таким образом, внутренняя энергия U является функцией двух переменных – энтропии (S) и объёма (V): U=f(S, V) Энтальпия H = U + pV; dH = dU + pdV + Vdp;
dH = TdS – pdV + pdV + Vdp = TdS + Vdp dH = TdS + Vdp
Частные производные энтальпии:
а) p=const; dp=0; dH=TdS; (H/S)p=T •
б) S=const; dS=0; dH= Vdp; (H/p)S= V •
Полный дифференциал функции H:
• dH = (H/S)p·dS + (H/p)S·dp Энтальпия H является функцией двух • переменных – энтропии (S) и давления (p):
H=f(S, p) Энергия Гельмгольца
• Для нахождения полного дифференциала функции F продифференцируем уравнение F = U – TS:
dF = dU – TdS – SdT (1)
• Q = dU + pdV, Q = TdS; TdS = dU + pdV, dU = TdS – pdV (2)
• После подстановки уравнения (2) в уравнение (1) получим:
• dF = TdS – pdV – TdS – SdT = – SdT – pdV, dF = – SdT – pdV (3)
• Из уравнения (3) находим частные производные (F/T)V = -S; (F/V)T = -p, dF = (F/V)T·dV + (F/T)V·dT
• Энергия Гельмгольца F является функцией двух переменных – объёма (V) и температуры (T): F=f(V, T) Энергия Гиббса
•Для нахождения полного дифференциала функции G продифференцируем уравнение G = H – TS:
dG = dH – TdS – SdT (1) H = U + pV; dH = dU + pdV + Vdp (2)
•После подстановки уравнения (2) в уравнение (1) получим:
dG = dU + pdV + Vdp – TdS – SdT (3)
•Из объединенного уравнения 1-го и 2-го законов термодинамики имеем: dU = TdS – pdV (4)
•После подстановки уравнения (4) в уравнение (3) получим:
dG = TdS – pdV + pdV + Vdp – TdS – SdT, dG = Vdp – SdT (5)
•Из уравнения (5) находим частные производные (G/p)T = V; (G/T)p = — S dG = (G/p)T·dp + (G/T)p·dT
•Энергия Гиббса G является функцией двух переменных – давления (p) и температуры (T): G=f(p, T)
• Следует отметить, что все вышеприведенные соотношения справедливы только для закрытых систем постоянной массы и состава.
Они применимы, например, для процессов фазовых превращений (испарение, плавление, кристаллизация и т.д).
• Для открытых же систем, в которых масса и состав могут изменяться за счёт выведения или добавления вещества, или за счёт протекания химической реакции, внутренняя энергия U, энтальпия H, энергия Гельмгольца F и энергия Гиббса G являются также функцией количества образующих данную систему индивидуальных компонентов.
Уравнения Гиббса-Гельмгольца Уравнения Гиббса-Гельмгольца – это термодинамические соотношения, устанавливающие связь между внутренней энергией (U) и энергией Гельмгольца (F), или между энтальпией (H) и энергией Гиббса (G):
F = U + T(F/T)V (1) G = H + T(G/T)p (2) Эти уравнения выражают зависимость F и G от температуры для индивидуального вещества.
Уравнение (1) следует из определения энергии Гельмгольца (F = U – TS) и выражения для энтропии S = -(F/T)V Уравнение (2) – из определения энергии Гиббса (G = H – TS) и выражения для энтропии S = -(G/T)p
• Если система переходит из состояния 1 «реагенты»
в состояние 2 «продукты», то эти уравнения можно записать для изменений соответствующих потенциалов в результате протекания химической реакции:
F = U + T(F/T)V (3) = H + T(G/T)p (4) G
• Эти уравнения также известны под названием «уравнения Гиббса-Гельмгольца» для процессов.
Они позволяют получить зависимость F и G от T.
Преобразуем уравнение (4) в более явную и удобную форму. Сделаем перегруппировку и разделим обе части на T2 [(G/T)p]/T — G/T2 = — H/T2 [(G/T)/dT]p = -H/T2 • 1) H 0, [(G/T)/ T]p 0, значит с увеличением T растёт (G/T). Поскольку T всегда 0, то G 0, то есть, с увеличением T G увеличивается (G становится менее отрицательной величиной – более положительной).
• 2) H 0, [(G/T)/ T]p 0, значит с увеличением T снижается (G/T). Поскольку T всегда 0, то G 0, то есть, с увеличением T G уменьшается (G становится менее положительной – более отрицательной величиной).
Фундаментальные уравнения для открытых систем.
Понятие о химическом потенциале До сих пор мы рассматривали случаи, в которых изменение энергии системы было следствием сообщения или отнятия теплоты или результатом совершения работы. В открытых же системах масса и состав могут изменяться.
Ясно, что появление в открытой системе некоторого количества вещества или удаление его из системы должно сказаться на запасе энергии системы. Поэтому энергия Гиббса, энергия Гельмгольца, энтальпия и внутренняя энергия являются также функцией количества образующих данную систему индивидуальных компонентов.
• Рассмотрим это на примере энергии Гиббса:
G = f(p, T, n1, n2, … nk), где n1, n2, … nk – это число молей 1, 2, … k-го компонента.
• Поэтому полный дифференциал энергии Гиббса dG будет складываться не только из изменения энергии Гиббса в результате изменения давления и температуры (на dp и dT, соответственно), но и изменения энергии Гиббса в результате изменения числа молей каждого компонента (1-го на dn1, 2-го на dn2, kго на dnk).
• dG = (G/p)T,n1,n2,…nk·dp + (G/T)p,n1,n2,…nk·dT + (G/n1)T,p,n2,…nk·dn1 + (G/n2)T,p,n1,n3,…nk·dn2 + (G/nk)T,p,n1,n2,…n(k-1)·dnk, либо
• dG = (G/p)T,n1,n2,…nk·dp + (G/T)p,n1,n2,…nk·dT + (G/ni)T,p,ji·dni (1) (индекс j i – постоянное количество всех dni компонентов, кроме ni) Обозначим частную производную энергии Гиббса по количеству i-го вещества при постоянных давлении, температуре и количествах всех остальных веществ (G/ni)T,p,ji = i и назовем химическим потенциалом i-го компонента в системе данного состава
• С учётом того, что (G/p)T = V, (G/T)p = -S, уравнение (1) принимает вид dG = -SdT + Vdp + idni
• Полные дифференциалы для остальных термодинамических потенциалов имеют следующий вид:
dF = -SdT – pdV + idni dU = TdS – pdV + idni dH = TdS + Vdp + idni
Таким образом, из приведенных выше уравнений следует:
i = (U/ni)S,V,nj = (H/ni)S,p,nj = (F/ni)T,V,nj = (G/ni)T,p,nj Таким образом, химический потенциал можно определить как изменение любого вида энергии, сопровождающее изменение количества вещества в системе.
•Химический i-го многокомпонентной системы равен частной производной от любого из термодинамических потенциалов по количеству этого компонента при постоянных значениях остальных термодинамических переменных, определяющих данный термодинамический потенциал.
важным является выражение,
•Особенно связывающее химический потенциал с энергией
Гиббса:
• Величина i представляет собой бесконечно малое изменение энергии Гиббса, которое происходит в результате изменения количества молей данного iго компонента на бесконечно малую величину dni при постоянных давлении, температуре и числах молей всех других компонентов, кроме i-го. Но, поскольку dni бесконечно мало, а числа молей всех других компонентов, кроме i-го, не меняются, то можно считать, что i-ый компонент добавляется в систему при постоянном составе системы. Часто химический потенциал i относят к 1 молю добавляемого компонента, отсюда можно сформулировать физический смысл химического потенциала.
Физический смысл химического потенциала
• Это изменение энергии Гиббса однородной многокомпонентной системы при добавлении к ней 1 моль данного компонента при постоянных давлении, температуре и составе системы (то есть, добавление должно происходить при бесконечно больших количествах всех компонентов, чтобы состав системы не изменился).
• Химический потенциал относится к числу вычисляемых, но не измеряемых на опыте параметров.
• Химический потенциал характеризует способность рассматриваемого компонента к выходу из данной фазы (путём испарения, растворения, кристаллизации и т.д.) или к выходу из данного состояния при химическом взаимодействим. В многофазных (гетерогенных) системах переход данного компонента может происходить самопроизвольно только из фазы, где его химический потенциал больше, в фазу, в которой его химический потенциал меньше.
Такой переход сопровождается уменьшением химического потенциала этого компонента в 1-й фазе и увеличением во 2-й.
В результате, разность между химическими потенциалами данного компонента в этих двух фазах уменьшается и при достижении равновесия химический потенциал компонента становится одинаковым в обеих фазах.
В любой равновесной гетерогенной системе химический потенциал каждого компонента одинаков во всех фазах.
Понятие химического потенциала имеет некоторую аналогию с понятием температуры. Как в состоянии теплового равновесия система должна иметь одинаковую температуру, так и при постоянных давлении и температуре в состоянии равновесия химические потенциалы каждого компонента должны быть одинаковыми по всей системе, в любой из имеющихся фаз.
Таким образом, в неравновесных системах любой компонент будет стремиться из состояния с более высоким химическим потенциалом в состояние с более низким потенциалом до тех пор, пока не установится равновесие.
Рассмотрим процесс таяния льда или кристаллизации воды В гетерогенной системе вода-лед при температуре выше 0°С химический потенциал воды ниже, чем химический потенциал льда. В результате, молекулы воды переходят из твёрдой фазы в жидкую – лёд тает.
Наоборот, при температуре ниже 0°С химический потенциал льда ниже, чем химический потенциал воды. При этом молекулы воды переходят из жидкой фазы в твёрдую – происходит процесс кристаллизации.
Рассмотрим диссоциацию слабой кислоты.
HA + Н2О Н3О+ + AВ процессе диссоциации концентрация недиссоциированных молекул снижается, а концентрации ионов растут. Поэтому химический потенциал НА снижается, а сумма химических потенциалов Н+ и А- растёт. Когда сумма химических потенциалов продуктов станет равной сумме химических потенциалов реагентов система достигнет состояния равновесия.
Таким образом, разность величин определяет направление химических реакций, фазовых превращений, диффузии веществ из одной фазы в другую.
Химическое равновесие. Принцип минимума свободной энергии. Закон действующих масс.
Константа химического равновесия и способы её выражения. Кинетический и термодинамический выводы выражения для константы равновесия.
Термодинамическим равновесием называется такое термодинамическое состояние системы, которое при постоянстве внешних условий не изменяется во времени, причём, эта неизменяемость не обусловлена каким-либо внешним процессом.
Принцип минимума свободной энергии:
«самопроизвольно могут протекать только те процессы, которые приводят к понижению свободной энергии системы; система приходит в состояние равновесия, когда свободная энергия достигает минимального значения»
• Протекание самопроизвольного процесса в системе сопровождается уменьшением свободной энергии системы (dG 0; dF 0).
• Очевидно, что рано или поздно система достигнет минимума свободной энергии.
Таким образом, минимальное значение энергии Гиббса или энергии Гельмгольца является условием термодинамического равновесия в закрытой системе:
T и p = const: G = 0; dG = 0 V и T = const: F = 0; dF = 0
• Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации Ci или парциальное давление Pi реагирующих веществ.
• Для некоторой реакции:
• Соответствующие константы равновесия выражаются следующим образом:
KC = (CcC · CdD) / (CaA · CbB) (1), KP = (PcC · PdD) / (PaA · PbB) (2).
• Закон действующих масс (закон ГульдбергаВааге):
• «В состоянии равновесия отношение произведения равновесных концентраций (парциальных давлений) продуктов реакции к произведению равновесных концентраций (парциальных давлений) исходных веществ есть величина постоянная при данной температуре»
Кинетический вывод закона действующих масс Экспериментально установлено, что во многих случаях скорость химической реакции прямо пропорциональна концентрации реагирующих веществ. Если между веществами А и В происходит обратимая реакция и при этом образуются вещества C и D.
aA + bB cC + dD, то скорость прямой реакции можно определить из следующего соотношения:
vпр = k1·CaA·CbB (1), где k1 – константа скорости реакции; CaA и CbB – концентрации веществ А и В в степенях, равных стехиометрическим коэффициентам в уравнении реакции, в данный момент времени.
По мере развития химической реакции концентрации веществ А и В уменьшаются, а веществ C и D – увеличиваются.
При этом скорость обратной реакции возрастает и определяется по уравнению vобр = k2·CcС·CdD (2), где k2 – константа скорости обратной реакции; CcC и CdD – концентрации веществ C и D в степенях, равных стехиометрическим коэффициентам в уравнении реакции, в данный момент времени.
До наступления равновесия суммарная скорость реакции равна разности скоростей прямой и обратной реакций.
Равновесие наступает в тот момент, когда k1·CaA·CbB = k2·CcС·CdD (3) или k1/k2 = (CcC · CdD) / (CaA · CbB) = KC (4), где KC – константа равновесия реакции, зависящая от природы веществ и температуры и не зависящая от концентрации реагирующих веществ.
Концентрации газообразных веществ можно заменить соответствующими парциальными давлениями, при этом константа равновесия обозначается Kp:
Kp = (PcC · PdD) / (PaA · PbB) (5) Константы KC и Kp для одной и той же реакции численно не равны, между ними существует следующая зависимость:
Kp = KC·(RT)n (6) или KС = Kp·(RT) -n, (7) где n – приращение числа молей газообразных веществ.
Термодинамический вывод выражения для константы химического равновесия dG = Vdp – SdT + i·dni (1) Из уравнения (1) следует, что при p = const (dp = 0) и T = const (dT = 0) dG = i·dni (2) и, следовательно, G = ni·i (3) Уравнение (3) утверждает, что общая свободная энергия системы представляет собой сумму числа молей каждого компонента, умноженных на свободную энергию моля компонента (т.е. химический потенциал) при указанной концентрации.
Легко можно установить зависимость i от концентрации iго компонента:
i = 0i + RTlnCi (4), где Ci – молярная концентрация i-го компонента, 0i соответствует химическому потенциалу растворенного вещества, который должен был бы наблюдаться в растворах с единичной концентрацией i-го компонента.
Рассмотрим равновесную обратимую химическую реакцию aA + bB cC + dD Можно утверждать, что в состоянии равновесия (p и T = const) суммарная энергия Гиббса продуктов реакции равна суммарной энергии Гиббса реагентов, поскольку в противном случае равновесие сдвигается в ту или иную сторону:
GA + GB = GC + GD (5) Подставляя уравнение (3) в уравнение (5) находим, что aA + bB = cC + dD (6) а после подстановки уравнения (4) в уравнение (6) получаем a[0A + RTlnCA] + b[0B + RTlnCB] = c[0C + RTlnCC] + d[0D + RTlnCD], или с0C + d0D — a0A — b0B = RT[-clnCC – dlnCD + alnCA + blnCB] (G0C + G0D) – (G0A + G0B) = — RT[lnCcC + lnCdD – lnCaA – lnCbB] G0продукты – G0реагенты = G0r = = -RTln[(CcC · CdD)/(CaA·CbB)] (7) Основной смысл уравнения (7) состоит в том, что поскольку левая часть уравнения постоянна(то есть, не зависит от концентрации), то и правая часть должна также не зависеть от общих концентраций всех веществ, участвующих в реакции.
Следовательно, для обозначения выражения в скобках в уравнении (7) можно ввести новую величину – константу равновесия:
Kравн = (CcC · CdD)/(CaA·CbB), (p и T = const) G0r = -RTlnKравн Уравнение изотермы химической реакции Это уравнение легко получить используя следующее соотношение:
• При постоянной температуре (SdT = 0):
• Для одного моля идеального газа V = RT / P и, следовательно, dG = RT dP / P = RT d (lnP) (3) •
• Интегрируя, получаем G2 = G1 + RTln (P2 / P1) (4) • Это уравнение позволяет, зная молярную энергию Гиббса идеального газа G1 при парциальном давлении P1, вычислить молярную энергию Гиббса G2 при парциальном давлении P2. Допустив, что состояние 1 является стандартным, а состояние 2 произвольным, уравнение (4) можно записать в виде:
Gi = G0i + RTln ai, (5) где G0i – стандартный изобарно-изотермический потенциал вещества i; ai– C/C0 – его активная концентрация (активность).
В соответствии с (5) энергия Гиббса произвольной химической реакции aА + bВ = lL + mМ равна:
При достижении равновесия (G = 0) уравнение (6)принимает вид
где – равновесные значения активных концентраций.
Выражение под знаком логарифма, представляющее собой отношение произведения равновесных активностей продуктов к произведению активностей исходных веществ в степенях их стехиометрических коэффициентов, называется константой равновесия:
(8) Подставив (8) в (6), получим уравнение, носящее название изотермы Вант-Гоффа:
(9) При определенных условиях активности реагентов могут быть заменены концентрациями (Kc) или парциальными давлениями ( Kp). Для газообразных веществ Kp и Kc связаны соотношением Kp = Kc (RT)n, где n – разность числа молей начальных и конечных газообразных реагентов.
Анализ уравнения изотермы
1) ln[(CeE · CdD)/(CaA·CbB)] lnK.
Следовательно, G0. В системе термодинамически возможно самопроизвольное протекание прямой реакции при указанных выше условиях.
2) ln[(CeE · CdD)/(CaA·CbB)] lnK.
Следовательно, G0. В этом случае в системе термодинамически возможно самопроизвольное протекание обратной реакции при указанных выше условиях.
3) ln[(CeE · CdD)/(CaA·CbB)] = lnK.
Следовательно, G=0. Данная система будет находиться в состоянии химического равновесия.
Из анализа уравнения изотермы химической реакции видно, что меняя начальные концентрации давления) (парциальные веществ, в принципе, можно менять и направление протекания реакции.
Уравнения изобары и изохоры химической реакции Уравнения изобары и изохоры химической реакции определяют зависимость константы равновесия от температуры при p=const или V=const, соответственно.
Запишем уравнение Гиббса-Гельмгольца G = H + T (G/T)p и уравнение изотермы химической реакции G = RTln([C]·[D]/[A]·[B]) – RTlnKх.р.
Возьмём соответствующие производные по температуре (G/T)p = Rln([C]·[D]/[A]·[B]) – RlnKх.р. – RT(lnKх.р./T)p Подставим последние два уравнения в уравнение Гиббса-Гельмгольца RTln([C]·[D]/[A]·[B]) – RTlnKх.р. = H + RTln([C]·[D]/[A]·[B]) – RTlnKх.р. – RT2(lnKх.р./T)p 0 = H — RT2(lnKх.р./T)p (lnKх.р./T)p = H/(RT2) – уравнение изобары (lnKх.р./T)V = U/(RT2) – уравнение изохоры Анализ уравнения изобары
• Рассмотрим случай, когда реакция проводится при p=1 атм. Тогда уравнение (lnKp/T)p = H/RT2 примет следующий вид:
• dlnKp/dT = H0/RT2 • 1) H0 0, следовательно, H0/RT2 0, то есть, при увеличении температуры (dT 0) величина lnKp, а следовательно, и Kp возрастает, а при уменьшении температуры Kp уменьшается.
• 2) H0 0, следовательно, dlnKp/dT 0, то есть, с ростом аргумента Т величина lnKp, а, значит, и Kp уменьшается.
• 3) H0 = 0; dlnKp/dT = 0. Функция lnKp, а также и Kp, не зависит от температуры, то есть, Kp=const при Тconst.
• Проведенный анализ является частным случаем применения принципа ЛеШателье, когда внешним, воздействующим на систему, фактором является температура (то есть, как влияет изменение температуры на смещение равновесия в случае эндо- и экзотермических реакций).
• Чтобы определить изменение константы равновесия при заданном изменении температуры, нужно проинтегрировать уравнение изобары:
• dlnKх.р. = [H/(RT2)]·dT, T2 K2
• dlnKх.р. = [H/(RT2)]·dT T1 K1
• Если полагать, что тепловой эффект реакции в данном диапазоне не зависит от температуры, то получим при H = const:
• ln(K2/K1) = (H0/R) · (1/T1 – 1/T2)
 «Московский ордена Ленина, ордена Октябрьской Революции и ордена Трудового Красного Знамени Государственный университет имени М.В.Ломоносова ГЕОЛОГИЧЕСКИЙ ФАКУЛЬТЕТ Кафедра КРИСТАЛЛОГРАФИИ И КРИСТАЛЛОХИМИИ КУРСОВАЯ РАБОТА Расчет структуры и дефектов форстерита в ионном и ионноковалентном приближениях Студен. »
«Московский ордена Ленина, ордена Октябрьской Революции и ордена Трудового Красного Знамени Государственный университет имени М.В.Ломоносова ГЕОЛОГИЧЕСКИЙ ФАКУЛЬТЕТ Кафедра КРИСТАЛЛОГРАФИИ И КРИСТАЛЛОХИМИИ КУРСОВАЯ РАБОТА Расчет структуры и дефектов форстерита в ионном и ионноковалентном приближениях Студен. »
«А.П. Солодов Электронный курс 1 5 Компьютерное моделирование теплообмена: пакет Мatlab 5.1 Введение Математическое описание тепломассообмена (см. гл. 1–4 Электронного курса) включает систему дифференциальных уравнений. »
«ХИМИЯ РАСТИТЕЛЬНОГО СЫРЬЯ. 2009. №4. С. 59–62. УДК 661.728:66.095.112 СОРБЦИОННЫЕ И ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ХЛОПКОВОЙ ЦЕЛЛЮЛОЗЫ, ПОЛУЧЕННОЙ ИЗ РАЗЛИЧНОГО ХЛОПКОВОГО ЛИНТА Ф.И. Рузиев, М.Ю. Юнусов, А.А. Атах. »
«Лекция №13 Кристаллический мир в полиэдрах Теория плотнейших упаковок, ее использование для описания кристаллических структур Несмотря на многообразие современных приемов разбиения пространства, для описания атомного строения кристалла в кристаллохимии чаще всего. »
«ОСНОВНЫЕ НАПРАВЛЕНИЯ ИССЛЕДОВАНИЙ В БИОНЕОРГАНИЧЕСКОЙ ХИМИИ ВВЕДЕНИЕ Организм представляет сложным образом организованную систему, функционирование компонентов которой осуществляется в тесной взаимосвязи. Конечно, осн. »
«Министерство образования и науки Российской Федерации Государственное образовательное учреждение высшего профессионального образования «Ивановский государственный химико-технологический университет» Факультет неорганической химии и технологии Кафедра неорганической химии «УТВЕРЖДАЮ» Проректор по учебной работ. »
«IVANE JAVAKHISHVILI TBILISI STATE UNIVERSITY Mikheil Nodia Institute of Geophysics ТБИЛИССКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ им. И. ДЖАВАХИШВИЛИ Институт геофизики им. М. З. Нодиа. LXVI TRANSACTIONS vol. LXVI СБ. »
«МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РФ САРАТОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ им. Н.Г. ЧЕРНЫШЕВСКОГО М.В. Калинникова, Б.А. Головин, К.Б. Головин УЧЕБНОЕ ПОСОБИЕ ПО ГЕОФИЗИЧЕСКИМ ИССЛЕДОВАНИЯМ СКВАЖИН САРАТОВ – 2005 УДК 550.83(075) К 17 Учебное пособие составлено в соответствии с государственном стандартом специальности «011200-геофизика». »
2017 www.pdf.knigi-x.ru — «Бесплатная электронная библиотека — разные матриалы»
Материалы этого сайта размещены для ознакомления, все права принадлежат их авторам.
Если Вы не согласны с тем, что Ваш материал размещён на этом сайте, пожалуйста, напишите нам, мы в течении 1-2 рабочих дней удалим его.
Фундаментальные уравнения термодинамики. Преобразования Лежандра.
Глава 4. фундаментальные уравнения термодинамики. характеристические функции.
Фундаментальные уравнения термодинамики. Преобразования Лежандра.
Термодинамика как наука о наиболее общих свойствах макроскопических систем, находящихся в состоянии термодинамического равновесия, и о процессах перехода между этими состояниями построена просто – опытным путем установлены два ее основных закона, а применение к ним математического аппарата позволяет получить очень важные термодинамические соотношения.
Основой математического аппарата термодинамики является объединенное уравнение первого и второго законов термодинамики илифундаментальное уравнение Гиббса, которое для обратимых процессов записывается в виде
 , (4.1)
, (4.1)
где все параметры относятся к системе; PdV – механическая работа расширения системы; δW* – полезная работа системы (сумма немеханических видов работы, см. раздел 2.3)
Для простых систем в случае обратимых процессов фундаментальное уравнение записывается в виде
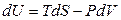 . (4.2)
. (4.2)
В математике независимыми переменными считаются те, которые стоят под знаком дифференциала. В фундаментальном уравнении (4.1) независимыми переменными являются S, V и все yk. Однако эти независимые параметры неудобны, так как энтропия непосредственно не измеряется, а объем легко определяется только для газов. Поэтому возникает задача перехода к новым независимым переменным.
Преобразование, меняющее ролями зависимые и независимые переменные, носит название преобразования Лежандра.
Рассмотрим функцию нескольких переменных:
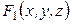 .
.
Полный дифференциал этой функции равен
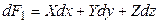 , (4.3)
, (4.3)
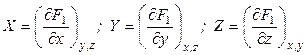 ,
,
Введем новую функцию
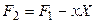 . (4.4)
. (4.4)
Полный дифференциал этой функции будет равен
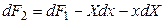 . (4.5)
. (4.5)
Подставив в равенство (4.5) значение dF1 из (4.3), получим:
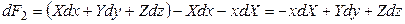 . (4.6)
. (4.6)
Cледовательно, в результате преобразования осуществлен переход от независимых переменных x, y, z к независимым переменным X, y, z, то есть переменная х стала зависимой, а Х – независимой. Кроме того, получили новую функцию F2. Таким образом, чтобы поменять зависимую переменную на независимую, следует воспользоваться соотношением
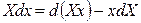 . (4.7)
. (4.7)
Впервые преобразование Лежандра к термодинамическим функциям применил Ф. Масье в 1869 году. Фундаментальное уравнение темодинамики для простых систем как для обратимых, так и для необратимых процессов запишется в виде:
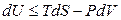 . (4.8)
. (4.8)
Знак неравенства используется для необратимых процессов, а знак равенства – для обратимых процессов. Применив к произведению PdV соотношение (4.7)
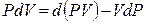 ,
,
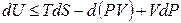 , (4.9)
, (4.9)
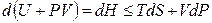 . (4.10)
. (4.10)
В результате перешли к независимым переменным S и Р и получили под знаком дифференциала в левой части (4.10) новую функцию
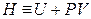 , (4.11)
, (4.11)
которая называется энтальпией.
Воспользовавшись подстановкой Лежандра для произведения TdS
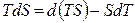 , (4.12)
, (4.12)
и подставив ее в фундаментальное уравнение (4.8), получим:
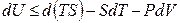 , (4.13)
, (4.13)
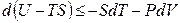 . (4.14)
. (4.14)
Стоящую под знаком полного дифференциала функцию U – TS обозначают по рекомендациям IUPAC символом Аи называютэнергией Гельмгольца(в некоторых учебниках энергию Гельмгольца до настоящего времени обозначают символом F):
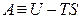 . (4.15)
. (4.15)
Итак, соотношение (4.14) записывается следующим образом:
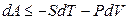 , (4.16)
, (4.16)
то есть в результате преобразований введена новая функция состояния при независимых переменных Т и V.
Преобразовав по Лежандру сразу оба произведения TdS и PdV в уравнении (4.8), получим:
 , (4.17)
, (4.17)
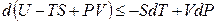 . (4.18)
. (4.18)
Функция U – TS + PV обозначается символом Gи называется энергией Гиббса. Следовательно, соотношение (4.18) запишется в виде:
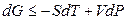 , (4.19)
, (4.19)
и энергия Гиббса является функцией независимых переменных Т и Р.
Дата добавления: 2015-08-20 ; просмотров: 3135 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Объединенное уравнение первого и второго начала термодинамики для физических и химических процессов
Объединенное уравнение первого и второго начала термодинамики, для равновесного обратимого процесса может быть выражено:
TdS = dU + pdV (4 – 1)
Отсюда следует, что
dU = TdS – pdV (4 – 1)
Введя новые функции состояния
U ≡ U + pV , G ≡ H – TS , F ≡ U — TS
можно получить четыре эквивалентные формы записи:
 dU = TdS – pdV
dU = TdS – pdV
dF = — pdV – SdT (4 — 2)
Все выражения (4 – 2) справедливы лишь для закрытых систем, в которых не совершаются другие виды работ за исключением механической работы. В этом случае, как уже отмечалось
U = U ( S , V ) H = H ( S , P ) G = G ( P , T ) F = ( V , T ) являются термодинамическими потенциалами и определяют направления самопроизвольного протекания процесса. Но, совершенно очевидно, что эти процессы связаны либо с выделением энергии в форме теплоты, либо с совершением механической работы. Никакие другие процессы здесь не рассматриваются.
Если в системе протекает химическая реакция, она становится аналогом открытой системы, поэтому возникает необходимость учитывать обмен энергией и массой с окружающей средой. Поэтому термодинамические потенциалы будут функцией всех веществ участвующих в реакции и систему уравнений (4 – 2) следует записать:





Все производные по количеству вещества равны между собой и характеризуют приращение соответствующего термодинамического потенциала при измерении данного вещества при фиксированных естественных переменных и неизменных количествах остальных веществ: (4 — 4)
µ i = (  )S,V, nj≠i = (
)S,V, nj≠i = (  ) S,P,nj≠i = (
) S,P,nj≠i = (  ) V,T,nj≠i = (
) V,T,nj≠i = (  )P,T,nj≠i
)P,T,nj≠i
Эта новая функция, введенная Гиббсом, получила название химического потенциала. Химический потенциал фактически является движущей силой при массопереносе. Система самопроизвольно переходит в состояние с более низшим потенциалом. Более нагретое тело, с большей температурой, нагревает менее нагретое тело, с меньшей температурой, т.е. температура – потенциал при теплопередачи, т.е. температура выступает здесь движущей силой. Работа расширения совершается аналогично за счет уменьшения давления: давление потенциал в случае механической работы. Все потенциалы, как известно интенсивные переменные, т.е. не зависящие от массы. Любой самопроизвольный процесс – это процесс приводящий к выравниванию потенциалы в результате чего достигается равновесие.
Очевидно, что если в системе совершаются другие виды работ, связанные с переносом массы, то учет ∑μ idni не достаточен.
Уравнения (4 -3) справедливы лишь в том случае, если химическая реакция протекает в гомогенной системе и системе не совершает других видов работ, кроме механической. Очевидно в этом случае:
 dU =
dU =  μidni (S,V = const)
μidni (S,V = const)
dH = μidni (S, p = const)
dG = μidni (T, p = const)
dF = μidni (T,V = const)
Для гетерогенной системы эти уравнения должны быть записаны для каждой фазы в отдельности. При этом, равновесие в гетерогенной системе наступает лишь тогда, когда химические потенциалы всех компонентов во всех фазах выравниваются.
Среди рассмотренных термодинамических потенциалов в конкретных термодинамических расчетах наибольшее значение имеют энергия Гельмгольца F и энергия Гиббса G . Их естественные переменные наиболее удобны, поскольку легко реализуемы. Изменение этих функций в каком – либо процессе при постоянстве естественных переменных равно максимальной работе, которая система может совершить в этом процессе при проведении процесса обратимо. Изменение этих термодинамических потенциалов можно найти из термодинамических констант, поэтому они позволяют определить возможность самопроизвольного процесса и глубину его протекания:
∆ GT = ∆ HT — T ∆ ST (4 – 6)
∆ FT = ∆ HT — T ∆ ST (4 – 7)
Если условия протекания процесса не сильно отличаются от стандартных, то в расчете можно использовать термические константы, отнесенные к стандартным условиям. В этом случае расчет ∆ rG ° T может быть осуществлен:
где ∆ rH º T и ∆ rS º T рассчитываются на основе стандартных энтальпий образования и абсолютных энтропий с пересчетом к температуре T .
Изменения энергии Гиббса может быть рассчитано аналогично тепловому эффекту реакции с использованием энергий Гиббса образования:
∆ rG ° T = ∑ υi ∆ fG ° T — ∑ υj ∆ fG ° T (4 – 8)
Рассмотрим равновесия типа:

 Жидкость Пар
Жидкость Пар
Твердое тело Пар

 Твердое тело Жидкость
Твердое тело Жидкость
Равновесие, как известно, характеризуется равенством энергий Гиббса для индивидуального вещества.
Исходя из (4 – 2) для жидкой и паровой фаз мы можем записать:
 dG ж = V ж d р – S ж dT
dG ж = V ж d р – S ж dT
dG пар = V пар d р — S пар dT (4 – 9)
Если энергии Гиббса отнесены к 1 молю, то условие равновесия может быть записано:
Отсюда следует, что
Из системы (4 – 9) получаем:
V ж dp – S ж dT = V п dp – S п dt
Отсюда следует, что
 (4 – 10)
(4 – 10)
Уравнение (4 – 10) – это уравнение Клаузиуса – Клайперона, которое показывает, как изменяется давление с ростом температуры при изменении агрегатного состояния вещества.
Уравнение (4 – 10) применимо к любому фазовому переходу I рода. Оно может быть преобразовано с учетом того, что
∆ G ф.п. = ∆ H ф.п . — Т∆ S ф.п. = 0
Для процессов испарений и сублимации V тв/ж ›› V п. Считая, что пар подчиняется закону идеальных газов: V п = RT / P . Уравнение (4 – 10) можно преобразовать:
 (4 — 10′)
(4 — 10′)
Уравнения (4 – 10) и (4 – 10 ′ ) позволяют определить энтальпию фазового перехода по температурной зависимости давления паров. В случае плавления требуются дополнительные сведения о больших объемах жидкой и твердой фаз.
Рассмотрим несколько примеров таких расчетов.
Приняв энтальпию плавления льда при 273,15 К равный 333,5 Дж∙2ˉ¹, а удельные объемы жидкой воды и льда равными 1,0002 см³∙2ˉ¹ и 1,0908 см³∙2ˉ¹ соответственно, рассчитать на сколько нужно повысить давление, чтобы температура плавления льда уменьшилась на 1 К.
Из уравнения (4 – 10) следует 


Знак «-» указывает, что температура плавления льда уменьшается с ростом давления.
При давлении 1 атм. Бензол кипит при 80,1º С. Какое давление паров бензола при стандартной термодинамической температуре?
На основании уравнения (4 — 10′) получаем
 │
│ 
В условии задачи отсутствует ∆Н исп . Её можно найти через энтальпии образования, предположив, что энтальпия испарения слабо зависит от температуры.
 = 82,98 кДж∙мольˉ¹
= 82,98 кДж∙мольˉ¹
 = 49,07 кДж∙мольˉ¹
= 49,07 кДж∙мольˉ¹
Следовательно, ∆ бар Нº(298, С6Н6) = 33,91 кДж∙мольˉ¹


Возможна и другая оценка энтальпии испарения. Для неполярных жидкостей выполняется правило Трутона, согласно которому мольная энтальпия испарения в точке НТК (нормальной температуры кипения) примерно равна 88 Дж∙мольˉ¹ Кˉ¹.
Оценивая для бензола ∆Н исп., получаем ∆Н исп. = 88∙353,3 = 31100 Дж∙мольˉ¹. В этом случае получаем р=0,141 атм.
Какое давление паров бензола при стандартной термодинамической температуре, если стандартные энергии Гиббса образования жидкого и газообразного бензола равны +124,43 кДж∙мольˉ¹ и +129,75 кДж∙мольˉ¹ соответственно.
Если Т = const = 298,15 то

Принимая ∆ V = V пар = получаем, после интегрирования



При каком давлении алмаз и графит находятся в равновесии друг с другом при температуре 298,15 К, если плотность графита и алмаза 2,25 и 3,51 г∙смˉ³ соответственно, а  = 2,900 кДж∙мольˉ¹
= 2,900 кДж∙мольˉ¹
 Сграфит Салмаз
Сграфит Салмаз

∆ fG =2,900 кДж∙мольˉ¹
Поскольку  , то
, то 
По условию равновесие соответствует ∆G2 = 0, следовательно
P 2 – P 1 = ∆G2 — 
∆ G 1= — ∆ fG º. Если принять, что стандартные условия отнесены к Р1 = 1 атм. = 101325,1 Па,
то  =
= 
При 70ºС давление насыщенных паров CCl 4 621,15 мм рт. ст, а при 80ºС 843,29 мм рт. ст. Чему равна энтальпия испарения СС l .
Из уравнения Клаузиуса – Клайперона
 , получаем
, получаем
 │=
│=  │=
│= 
http://helpiks.org/4-93290.html
http://www.trotted.narod.ru/physchem/lec-4.htm