Реакции третьего порядка
При равных начальных концентрациях реагентов в общем виде кинетическое уравнение реакции третьего порядка
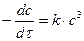 или –dc/с 3 = k·dτ.
или –dc/с 3 = k·dτ.
После интегрирования получаем:
где В – постоянная интегрирования.
При τ = 0 В = 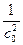 . Тогда
. Тогда
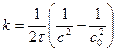 . (94)
. (94)
Константу скорости реакции третьего порядка измеряют в дм 6 /(моль 2 ·время).
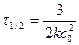 . (95)
. (95)
Для реакции третьего порядка период полупревращения обратно пропорционален квадрату начальной концентрации реагентов.
Порядок реакции определяют несколькими способами, используя опытные данные об изменении концентрации реагирующих веществ со временем.
1. Метод подстановки. Экспериментально находят концентрацию одного из веществ через определённые промежутки времени от начала реакции. Подставляют найденные значения концентрации в уравнения (88), (90), (92), (94) и рассчитывают значения константы скорости. Уравнение, дающее одинаковые значения константы скорости в различные моменты времени, указывает на порядок реакции.
2. Графический метод. Из приведённых уравнений следует, что зависимость концентрации от времени для различных порядков может быть выражена прямой линией в соответствующей системе координат: с – τ (для n=0), lnс – τ (для n=1), 1/с – τ (для n=2), 1/с 2 – τ (для n=3). Отложив на оси абсцисс время τ, а на оси ординат – с, lnс, 1/с, 1/с 2 , для изучаемой реакции получают четыре линии. Система координат, в которой экспериментальные данные лягут на прямую линию, укажет на порядок реакции, а тангенс угла наклона прямой к оси абсцисс равен константе скорости k (дляn=0,1,2) или 2 k (для n=3).
3. По периоду полупревращения. Период полупревращения для реакций различного порядка по-разному зависит от начальной концентрации реагентов (уравнения (89). (91), (93), (95)). Для реакции нулевого порядка τ1/2 прямо пропорционален начальной концентрации с0, для реакции первого порядка – не зависит от начальной концентрации с0, для реакции второго порядка – обратно пропорционален с0, для реакции третьего порядка – обратно пропорционален с 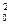 . Проводят опыты с различными начальными концентрациями реагентов, находят τ1/2 и делают вывод о порядке реакции.
. Проводят опыты с различными начальными концентрациями реагентов, находят τ1/2 и делают вывод о порядке реакции.
Дата добавления: 2014-12-07 ; просмотров: 3625 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
ФИЗИЧЕСКАЯ ХИМИЯ Химическая кинетика и катализ.
Химическая кинетика и катализ
Химическая кинетика – это учение о химическом процессе, его механизме и закономерностях протекания во времени. Химическая кинетика позволяет предсказывать скорость химических процессов. Создание математической теории химического процесса является в настоящее время необходимым условием для проектирования химических реакторов.
Содержание химической кинетики составляют два основных раздела:
а) формально математическое описание скорости реакции без учета механизма самой реакции ( математическое выражение процесса в целом), так называемая формальная (феноменологическая) кинетика;
б) учение о механизме химического взаимодействия на основе молекулярных данных о свойствах частиц – теории кинетики (молекулярная кинетика).
Скорость процесса химического взаимодействия зависит от условий, в которых протекает реакция. В соответствии с тем, в какой фазе протекает реакция, различают кинетику газовых реакций, кинетику реакций в растворах и кинетику реакций в твердой фазе.
В системах, в которых протекает последовательно несколько процессов, скорость всего процесса в целом определяется наиболее медленной, так называемой определяющей (лимитирующей) стадией. Если лимитирующей стадией является сам акт химического взаимодействия и процесс подвода и отвода компонентов реакции не влияет на ее скорость, то говорят, что реакция протекает в кинетической области. Здесь величина скорости реакции определяется кинетическим законам собственно химической стадии процесса. Могут встречаться реакции, в которых определяющей стадией является подвод реагирующего вещества, а не сам акт химического взаимодействия. В этом случае процесс протекает в диффузионной области, и скорость всего процесса определяется законами диффузии. Кроме того, бывают случаи, когда скорости реакции и диффузии соизмеримы. Скорость всего процесса тогда является сложной функцией кинетических и диффузионных явлений, и процесс протекает в переходной области.
Кинетическая классификация реакций
1. По числу частиц, участвующих в реакции:
2. По природе частиц, участвующих в элементарном акте реакции:
· реакции, в которой участвуют молекулы, называются молекулярными;
· реакции с участием свободных радикалов или атомов называются цепными;
· реакции с участием ионов называются ионными.
3. По числу фаз, участвующих в реакции:
· Гомогенными называются реакции, протекающие в однородной среде (например, в смеси реагирующих газов или в растворе);
· Гетерогенными называются реакции, протекающие в неоднородной среде, на поверхности соприкосновения взаимодействующих веществ, находящихся в разных фазах (например, твердой и жидкой, газообразной и жидкой и т.п.).
4. По степени сложности:
В зависимости от механизма реакции могут быть подразделены на простые и сложные. К простым реакциям относятся реакции, протекающие в одном направлении и включающие один химический этап. Формально они классифицируются по порядку (когда стехиометрический и кинетический порядки совпадают) и могут быть нулевого, первого, второго, третьего порядков. В «чистом виде» простые реакции почти не встречаются. В подавляющем большинстве случаев это стадии сложных химических процессов.
Сложные реакции – это многостадийные химические процессы. В кинетике существует положение о независимом протекании отдельных стадий сложной реакции: значение константы скорости отдельной стадии не зависит от наличия в системе других стадий.
Сложные реакции подразделяются следующим образом:
— обратимые реакции: А + В  Х + У
Х + У
В случае параллельных реакций скорость всего процесса будет определяться скоростью наиболее быстрой его стадии.
Если в последовательных реакциях скорость одной из химических стадий значительно меньше скоростей других, то скорость всего процесса будет определяться скоростью самой медленной стадии.
1.1. Понятие о скорости химической реакции
Для гомогенной химической реакции, протекающей при постоянном объеме, скоростью процесса по некоторому веществу называется изменение концентрации этого вещества в единицу времени. Если реакция описывается стехиометрическим уравнением:
Скоростью реакции называется величина:
 w = –
w = – 
 . (1.2)
. (1.2)
В кинетике принимают, что w > 0, поэтому знак «минус» в формуле (1.2) стоит потому, что сама производная отрицательна. Кроме того, скорость данной реакции имеет одинаковое значение независимо от того, через изменение концентрации какого из реагентов она выражена.
Скорость реакции зависит от природы веществ, их концентрации, температуры, наличия катализаторов и других факторов. Установление по экспериментальным данным вида уравнения зависимости скорости реакции от концентрации (кинетического уравнения) составляет одну из задач феноменологической кинетики. Для простых или элементарных реакций (односторонних, одностадийных химических процессов), а также для элементарных стадий сложных реакций эта зависимость устанавливается законом действующих масс, сформулированным норвежскими учеными Гульдбергом и Вааге (1864-1867): скорость реакции пропорциональна произведению концентраций реагентов (исходных), возведенных в степени, равным абсолютным значениям стехиометрических коэффициентов.
Если реакция (1.1) является элементарной, то основное кинетическое уравнение ее скорости (математическое выражение закона действующих масс) запишется в виде:
В некоторых случаях для сложных реакций справедливо выражение типа:
которое различается с (1.3) тем, что  могут отличаться от стехиометрических коэффициентов.
могут отличаться от стехиометрических коэффициентов.
Показатель степени при концентрации CАi в кинетическом уравнении скорости реакции называют порядком реакции по веществу Аi. Различают порядки стехиометрические  и кинетические
и кинетические  . Для простых реакций порядки совпадают:
. Для простых реакций порядки совпадают:  =
=  . В случае сложных реакций порядки могут быть равны, а могут быть и неравны между собой (признак сложной реакции). Порядок реакции может быть целым (
. В случае сложных реакций порядки могут быть равны, а могут быть и неравны между собой (признак сложной реакции). Порядок реакции может быть целым ( ,
, ) или дробным (
) или дробным ( ), положительным (
), положительным ( ,
, ) или отрицательным (
) или отрицательным ( ).
).
Общим или суммарным порядком реакции n называется сумма показателей степеней концентраций в основном кинетическом уравнении скорости реакции.
В кинетическом уравнении скорости реакции коэффициент пропорциональности k, не зависящий от концентрации, называется константой скорости или удельной скоростью, то есть скоростью, отнесенной к единице концентрации. Ее величина изменяется в широких пределах в зависимости от рода реакции и быстро растет с повышением температуры.
Формально простые кинетические уравнения типа (1.3) обычно представляют собой интерполяционные формулы сложных химических реакций. Одной из причин получения дробных порядков может быть тот факт, что реакция идет по нескольким путям при получении одного и того же продукта.
Важная характеристика реакции – ее молекулярность.
Для простой реакции молекулярность – это число частиц, принимающих участие в элементарном акте химического взаимодействия. В данном случае порядок и молекулярность совпадают. Простые реакции могут быть мономолекулярными, бимолекулярными, тримолекулярными. Участие в элементарном акте более трех частиц маловероятно.
В сложных реакциях конечный продукт образуется в результате протекания нескольких стадий. В этих случаях под молекулярностью понимают число частиц, испытывающих химическое превращение в соответствии со стехиометрическими уравнением реакции. Здесь порядок и молекулярность могут не совпадать.
Кинетика реакций первого порядка
В общем виде можно записать:
Пусть в исходный момент времени имеется а моль исходного вещества А. К моменту времени t прореагировало х моль вещества и осталось а – х моль вещества А.
Основное кинетическое уравнение скорости реакции первого порядка будет иметь вид:
Разделим переменные и проинтегрируем:
Получаем уравнение для расчета константы скорости:
k =  . (1.6)
. (1.6)
Размерность константы скорости первого порядка [время -1 ].
Константу скорости можно найти также графически:





На рисунке показаны зависимости х=f(t) и (а–х)=f(t). В точке пересечения а–х=х, значит х=а/2, т.е. к моменту времени t1/2 прореагирует половина взятого вещества, t1/2 называется временем или периодом полупревращения.

Период полупревращения реакций первого порядка:
Кинетика реакций второго порядка
Стехиометрическое уравнение реакции можно записать в общем виде
Если а и b – начальные мольные концентрации реагирующих веществ А и В, а х – число моль в 1 л, которое прореагировало за время t, то основное кинетическое уравнение скорости реакции второго порядка можно представить:
Интегрирование приводит к уравнению константы скорости (а b):
b):
k =  (1.9)
(1.9)
В простейшем случае концентрации веществ А и В одинаковы и а = b.
При этих условиях основное кинетическое уравнение скорости реакции второго порядка имеет вид:
Интегрирование дает уравнение константы скорости (а = b):
k =  . (1.11)
. (1.11)
Для определения значения k можно также использовать графический метод:
В случае а = b строится график в координатах 1/(а – х) = f(t), константа скорости равна: k = tg a .



Значение k для реакции второго порядка зависит от единиц, в которых выражена концентрация. Если выразить концентрацию в моль/л, а время в с, тогда константа скорости реакции второго порядка имеет размерность
Период полупревращения реакций второго порядка:

Кинетика реакций третьего порядка
Для реакции 3-го порядка вида А + В + С ® продукты
можно написать следующее основное кинетическое уравнение:
В частном случае, когда a=b=c, основное кинетическое уравнение скорости реакции третьего порядка имеет вид:
В результате интегрирования получим уравнение константы скорости:
k =  . (1.14)
. (1.14)
Для графического нахождения константы скорости строят зависимость:

Размерность константы скорости таких реакций [л 2 . моль -2 . с -1 ].
Период полупревращения реакций третьего порядка:

Кинетика реакций нулевого порядка
Существуют реакции, скорость которых не меняется с изменением концентрации одного или нескольких реагирующих веществ, поскольку она определяется не концентрацией, а некоторым другим ограничивающим фактором, например, количеством поглощенного света при фотохимических реакциях или количеством катализатора в каталитических реакциях. Тогда основное кинетическое уравнение скорости реакции имеет вид:
Константа скорости реакции нулевого порядка:

1.3. Методы определения порядка реакции
Все методы определения порядка реакции можно разделить на интегральные и дифференциальные.
1) Метод подстановки.
Заключается в том, что подстановка экспериментальных данных в уравнения нулевого, первого, второго, третьего порядков должна в одном из случаев дать постоянное значение константы скорости.
2) Графический метод.
График, построенный по опытным данным для концентраций рассматриваемого исходного вещества в разные моменты времени протекания реакции, будет выражаться прямой линией в разных координатах в зависимости от порядка данной реакции по веществу:



3) По периоду полупревращения.
Получают экспериментальные данные по τ½ при разных начальных концентрациях а, затем их анализируют.
Реагирующие вещества берут в одинаковых концентрациях. Определяют скорость реакции при двух концентрациях в различные промежутки времени:

 а – х1
а – х1


 а – х2 α2
а – х2 α2
 |
 |




2) По периоду полупревращения.
Для двух различных начальных концентраций а1 и а2 период полупревращения различен, т.к. он обратно пропорционален начальной концентрации в степени (n – 1).
Таким образом, определив по экспериментальным кривым «концентрация – время» период полупревращения для двух различных начальных концентраций:



 а – х а – х
а – х а – х

 а1 а2
а1 а2




 а1/2 а2/2
а1/2 а2/2

можно вычислить порядок реакции:
 при n ≠ 1,
при n ≠ 1,


2.1. Влияние температуры на скорость реакции
Обычно при повышении температуры скорость химической реакции увеличивается. Из этого общего правила известны лишь несколько исключений (например, реакция третьего порядка: 2 NO + O2 ® 2 NO2).
Влияние температуры на скорость химической реакции количественно может быть охарактеризовано в узком интервале температур величиной температурного коэффициента скорости реакции.
Температурным коэффициентом скорости реакции называется отношение констант скоростей реакции при двух температурах, отличающихся на 10 0 :
g = 
g =  (2.1)
(2.1)
Для подавляющего большинства реакций температурный коэффициент больше 1, причем величина его может изменяться в широких пределах. У многих реакций в растворах, при комнатной температуре, а также у ряда реакций в газовой фазе, протекающих при более высоких температурах, согласно эмпирическому правилу Вант-Гоффа, повышение температуры на 10 0 вызывает возрастание скорости реакции в 2  4 раза.
4 раза.
Объяснение больших величин температурного коэффициента дано теорией активных соударений Аррениуса.
Большие величины температурного коэффициента скорости реакции, а также несоответствие между числом столкновений реагирующих молекул и скоростью реакции позволили Аррениусу сформулировать его теорию активных соударений (ТАС)
Согласно Аррениусу, всякая реакция протекает через промежуточную стадию, связанную с образованием активных молекул:
Нормальные молекулы  активные молекулы ®продукты реакции.
активные молекулы ®продукты реакции.
Уравнение (2.2) является теоретическим обоснованием эмпирического правила Вант-Гоффа и объясняет большие величины температурного коэффициента.
 (2.2)
(2.2)
где k – экспериментально определяемая константа скорости реакции;
k0 – предэкспоненциальный множитель.
Еа – тепловой эффект перехода нормальных молекул в активные, так называемая энергия активации.
Энергия активации – это та энергия, которую должны накопить нормальные молекулы, чтобы иметь возможность прореагировать.
Количественным критерием возрастания скорости реакции (константы скорости) с повышением температуры является величина энергии активации, поэтому множитель е -Еа/ RT в уравнении (2.2) называют фактором активации.
В дифференциальной форме уравнение (2.2) имеет вид:
 (2.3)
(2.3)
Зависимости (2.2, 2.3) представляют собой уравнения Вант-Гоффа –Арениуса. Они показывают зависимость константы скорости реакции от температуры. Из формул видно, что логарифм экспериментально определяемой константы скорости является линейной функцией обратной температуры при условии, что энергия активации не зависит от температуры:
ln k = –  + ln k0 (2.4)
+ ln k0 (2.4)
где ln k0 – постоянная интегрирования.
Уравнение (2.4) можно использовать для графических расчетов энергии активации по экспериментальным данным. Для этого необходимо определить константы скорости при нескольких температурах и отложить на графике ln k в функции обратной температуры.

Взяв определенный интеграл от уравнения (2.3), получим получим интегральную форму уравнения Аррениуса:
 (2.5)
(2.5)
Уравнение (2.5) можно использовать для аналитических расчетов энергии активации по экспериментальным данным:
 .
.

На рисунке представлено Максвелл-Больцмановское распределение молекул по энергиям. Молекулы, для которых Е  Еа (заштрихованная область), являются активными (реакционноспобными). Реакция протекает благодаря наличию активных молекул.
Еа (заштрихованная область), являются активными (реакционноспобными). Реакция протекает благодаря наличию активных молекул.

На рисунке показано изменение энергии реагирующей системы. Здесь области: I – исходные молекулы, II – активные молекулы, III – продукты реакции, Еа – энергия активации, DН – тепловой эффект реакции. Избыточная по сравнению со средним значением энергия необходима для разрыва или ослабления связей в молекулах реагирующих веществ. Таким образом, под энергией активации понимают минимальное значение суммарной энергии сталкивающихся молекул, которая обеспечивает вступление их в реакцию между собой.
Активирование осуществляется путем соударения молекул. Протекание реакции, возможность ее осуществления обусловлены столкновением активных частиц. Столкновения как неактивных, так и активных частиц, как правило, являются бинарными вследствие малой вероятности тройных соударений. Теория активных столкновений основана на двух предпосылках:
1) наличие в системе активных молекул, возникающих за счет столкновения неактивных по законам статистического распределения энергии;
2) осуществление реакции возможно только в результате столкновения активных молекул.
Под активными молекулами понимают частицы, обладающие избыточным запасом энергии, достаточным для преодоления энергетического барьера реакции.
Из рисунка следует, что скорость реакции, пропорциональная числу активных молекул, зависит от энергии активации. Уровень «активные молекулы» определяет тот наименьший запас энергии, которым должны обладать молекулы, чтобы их столкновения могли привести к химическому взаимодействию. Разность между данным и исходным уровнем представляет собой энергию активации прямой реакции Еа. Таким образом, по пути от исходного состояния в конечное система должна перейти через своего рода энергетический барьер. Только активные молекулы, обладающие в момент столкновения необходимым избытком энергии, могут преодолевать этот барьер и вступить в химическое взаимодействие.
В ряде случаев наблюдаемая скорость реакции гораздо меньше вычисляемой по уравнению (2.4). Для таких реакций в уравнение вводится поправочных множитель, который находится опытным путем.
где р – поправочный множитель, называемый фактором вероятности или стерическим (пространственным) фактором. Этот фактор должен учесть такие особенности во взаимодействии молекул, как, например, ориентацию молекул в момент столкновения, распад активных молекул до столкновения, «неудачные удары» и т.д.
1. Теория активных соударений рассматривает только результат соударения, но не его сам акт;
2. Теория Аррениуса дает возможность вычислить энергию активации реакции в целом, но не объясняет ее связи с механизмом реакции и строением молекул, не объясняет физического смысла предэкпоненциального множителя;
Теория активного комплекса (ТАК) представляет собой дальнейшее развитие теории активных соударений. Она детально изучает сам акт соударения, рассматривает его энергетику. Используя квантово-статистические методы, ТАК изучает энергетический процесс соударения, рассматривает физику активных соударений и химического превращения. Согласно этой теории, активные соударения, приводящие реакции, являются сложным процессом постепенного перераспределения связей в молекуле, который начинается еще до столкновения молекул и заканчивается только после того, как молекулы как молекулы разойдутся на расстояния, превышающие дальность действия их силовых полей. Теория активного комплекса основана на том, что элементарный акт взаимодействия молекул состоит в постепенной перестройке химических связей. В любом элементарном акте взаимодействия первой стадией является сближение молекул, приводящее к образованию активных групп или активного комплекса, который может либо вновь распадаться либо дать продукты реакции.
Например, реакцию между молекулярным водородом и йодом с образованием йодистого водорода схематически можно изобразить следующим образом:

 H J H — – —J H2 + J2
H J H — – —J H2 + J2

 | + | ® | | (2.6)
| + | ® | | (2.6)
 H J H— – —J 2HJ
H J H— – —J 2HJ
Приближение молекулы водорода к молекуле йода приводит к постепенному ослаблению связи между атомами в этих молекулах. В момент наибольшего сближения молекул связи между всеми атомами становятся равноценными и все атомы принадлежат одному (переходному) состоянию или состоянию активного комплекса. Активный комплекс не является молекулой или промежуточным соединением, потому что реагирующие молекулы в переходном состоянии обладают максимальной энергией. Здесь не может быть равновесного состояния. Реакцию (2.6) нужно рассматривать как одностадийную реакцию. Время жизни комплекса ничтожно мало
( с). Дальнейшее движение атомов приводит к уменьшению расстояния между ними во вновь образовавшейся молекуле HJ. Однако, возможна и обратная картина – распад образовавшегося активированного комплекса на исходные частицы.
с). Дальнейшее движение атомов приводит к уменьшению расстояния между ними во вновь образовавшейся молекуле HJ. Однако, возможна и обратная картина – распад образовавшегося активированного комплекса на исходные частицы.
Рассмотрим процесс взаимодействия молекулы АВ с молекулой С. Это взаимодействие происходит с обязательной промежуточной стадией – образованием активированного комплекса А – – В – – С по схеме:
Образование активного комплекса связано с ослаблением связей в молекулах реагирующих веществ, т.е. первоначально с затратой работы и соответственным увеличением потенциальной энергии системы. Таким образом, в процессе реакции происходит сначала рост, а затем падение потенциальной энергии системы. Исходное АВ + С и конечное А + ВС состояние системы разделены энергетическим барьером.
В ходе реакции расстояния между атомами в реагирующих молекулах изменяются, а в переходном состоянии они между всеми атомами становятся соизмеримыми. На рис.1 показано изменение потенциальной энергии показано изменение потенциальной энергии системы атомов АВС вдоль координаты пути реакции х: АВ + С – исходное состояние системы, А + ВС – конечное состояние.


Рисунок 1 Рисунок 2
Координата реакции х является величиной, характеризующей перемещение системы по ходу реакции, вдоль пути, наиболее выгодного энергетически. Эта величина всегда должна возрастать по ходу процесса. Таким образом, в ходе элементарного акта химического превращения система преодолевает энергетический процесс. Долины Р1 и Р2 разделяют энергетический барьер Р. Разность между потенциальной энергией Р1 исходных веществ и потенциальной энергией активного комплекса в перевалочной точке Р равна энергии активации. На рис. 2 показано изменение потенциальной энергии при перемещении вдоль координаты реакции Р1РР2. Здесь вместо пространственного изображения применена топографическая схема с использованием линий, показывающих эквипотенциальные поверхности.
Термодинамическое обоснование теории активного комплекса
Статистическая термодинамика дает наиболее общее уравнение теории активного комплекса:
kск =  æ
æ  (2.7)
(2.7)
где h – постоянная Планка, 6,62 . 10 -34 Дж . с;
k – постоянная Больцмана, 1,38 . 10 -23 Дж/К;
К * – константа равновесия между активными комплексом и исходными веществами;
æ – трансмиссионный коэффициент или коэффициент прохождения. Он учитывает долю активных комплексов, скатывающихся с перевала Р в долину Р2 и превращается в конечные продукты). Для большинства реакций æ близок к 1.
Из 2 закона термодинамики:  G * =
G * =  H * – T
H * – T S * .
S * .
Для равновесия между активным комплексом и исходными веществами:
 G * = —RT lnK *
G * = —RT lnK *
 ;
; 
Подставляем в (2.7):
kск = æ
 (2.8)
(2.8)
Из уравнения (2.8) можно вычислить kск, зная  H * и
H * и  S * , где
S * , где  S * – изменение энтропии активации. Физический смысл ее определяет долю столкновений, когда молекулы ориентированы надлежащим образом.
S * – изменение энтропии активации. Физический смысл ее определяет долю столкновений, когда молекулы ориентированы надлежащим образом.
Чтобы выяснить физический смысл  H * , прологарифмируем уравнение (2.7):
H * , прологарифмируем уравнение (2.7):
ln kск = ln æ  + lnT + lnK *
+ lnT + lnK *
Дифференцируем по Т: 
Из теории Аррениуса: 
Из уравнения изобары Вант-Гоффа: 
Значит,  ,
,
Для большинства реакции Еа >>RT (Ea  50 – 200 кДж/моль; при 298 К, а RT = 2,5 кДж/моль). Поэтому величиной RT в уравнении (2.9) можно пренебречь и считать ΔH *
50 – 200 кДж/моль; при 298 К, а RT = 2,5 кДж/моль). Поэтому величиной RT в уравнении (2.9) можно пренебречь и считать ΔH *  Еа.
Еа.
Теория активного комплекса позволяет вычислить стерический фактор р. Приравнивая правые части уравнений (2.8) и уравнение Аррениуса, считая, что ΔH *  Еа, получим
Еа, получим
р = æ 
Кинетика гетерогенных реакций.
Гетерогенными называются реакции, протекающие на поверхности раздела фаз. Такие реакции могут протекать в двух областях: диффузионной и кинетической.
В кинетической области kреак > kдиф и скорость всего процесса определяется диффузионными зависимостями.
Отличительные особенности диффузионной области:
1) малые величины энергии активации – Едиф.обл. 10 ккал;
2) влияние перемешивания на скорость реакции.
Рассмотрим процесс диффузии.
Диффузией называется самопроизвольное перемещение вещества, приводящее к равномерному распределению концентраций в объеме.
Диффузия может осуществляться только в тех случаях, когда в различных точках пространства концентрация веществ различна. Движущей силой диффузии является градиент концентрации. Это изменение концентрации на отрезке пути dx –  .
.
Обозначим dm –количество вещества, проходящее при диффузии через площадь S, за время dt.
Тогда  Первый закон Фика.
Первый закон Фика.


 ;
; 
D – коэффициент диффузии [м 2 /с]
Коэффициент диффузии представляет собой количество вещества, проходящего в единицу времени через единицу площади, при градиенте концентрации равном единице.
D = f(температуры, природы вещества).
 Температурный коэффициент скорости диффузии, α ≈ 1,2, что объсняется малым значением энергии активации диффузионного процесса.
Температурный коэффициент скорости диффузии, α ≈ 1,2, что объсняется малым значением энергии активации диффузионного процесса.
Рассмотрим диффузионную кинетику реакции при стационарном состоянии диффузионного потока. Возьмем некоторый объем, в котором происходит диффузия.

Стационарное состояние диффузионного потока характеризуется тем, что в элемент объема dx в единицу времени входит такое же количество вещества, какое выходит из этого объема.
Скорость диффузии при этих условиях будет равна:

1) Рассмотрим случай, когда реакция протекает в диффузионной области.
Пусть С1 – концентрация вещества в объеме раствора;
С2 – концентрация вещества у поверхности твердой фазы.
Тогда : 
Обозначим  – экспериментальная константа скорости
– экспериментальная константа скорости

2) Рассмотрим кинетические зависимости в тех случаях, когда скорость диффузии и скорость химической реакции соизмеримы.
Примем S = 1м 2 , тогда

 константа скорости диффузии;
константа скорости диффузии;
δ – толщина диффузионного слоя.
Для химической реакции возьмем наиболее простой случай (n =1). В реакцию вступают только те вещества, компоненты которых находятся в поверхностном слое.

при установившемся процессе 

 концентрация вещества в поверхностном слое
концентрация вещества в поверхностном слое



Обозначим  – константа скорости реакции в смешанной области.
– константа скорости реакции в смешанной области.
Тогда:  Скорость гетерогенной реакции.
Скорость гетерогенной реакции.
Можно также записать:  ,
,
где  — диффузионное сопротивление.
— диффузионное сопротивление.
 – химическое сопротивление.
– химическое сопротивление.
3. К а т а л и з
3.1. Основные понятия катализа
Катализом называют явление увеличения скорости реакции, происходящее под действием некоторых веществ (катализаторов), которые, участвуя в процессе, остаются химически неизменными.
Имеются также вещества, которые наоборот уменьшают скорость реакции – ингибиторы. А явление называется ингибированием или отрицательным катализом.
Общий механизм каталитического действия состоит в том, что реагирующее вещество и катализатор образуют промежуточное соединение, которое реагирует с другим исходным веществом с образованием продуктов реакции и регенерации молекул катализатора.
Схема процесса: А + В = (АВ) * → С + D
Природа промежуточных соединений в катализе разнообразна. Чаще всего они представляют собой лабильные молекулы или радикалы, существующее лишь очень короткое время.
Если рассматривать катализ с энергетической точки зрения, то можно заметить, что катализатор ведет реакцию по иному пути, чем тот, который отвечает реакции без катализатора. Поэтому энергия активации каталитической реакции значительно ниже энергии активации реакции без катализатора. Т.к. энергия активации входит в показатель степени в уравнении для константы скорости, то даже сравнительно небольшое снижение энергии активации сильно увеличивает скорость химического превращения.
 Uпот (АВ) *
Uпот (АВ) *


 Ек * К 2. каталитическая реакция
Ек * К 2. каталитическая реакция


 Еа
Еа
 С+D
С+D



 ΔЕк = Еа – Ек
ΔЕк = Еа – Ек
3.2. Свойства катализаторов
В промышленности используются различные катализаторы, но все они обладают рядом общих свойств, которые присущи и самому явлению катализа в целом:
1. Катализаторы ускоряют течение только тех реакций, которые термодинамически возможны, т. е (ΔG + и ОН — .
5. при одновременном параллельном действии нескольких катализаторов или при параллельном образовании нескольких промежуточных соединений за счет одного катализатора общая скорость процесса равна сумме скоростей разложения отдельных промежуточных соединений.
Механизм гетерогенного катализа
На практике наиболее часто встречаются 2 типа гетерогенного катализа: а) катализатор находится в твердой фазе, а реагирующие вещества – в жидкой; б) катализатор находится в твердой фазе, а реагирующие вещества – в газообразной.
Пусть в отсутствии катализатора протекает реакция.
А + В = (АВ) * → продукты
Предположим, что активные состояния (АВ) * для каталитической и некаталитической реакции аналогичны.
Весь гетерогенно-каталитический процесс можно разделить на следующие стадии:
1) адсорбция исходных веществ на поверхности катализатора —
Этот процесс активированный и экзотермический, т.е. состояние АВК будет обладать меньшей потенциальной энергией по сравнению с (А+В+К)
2) перевод адсорбированного состояния в активное —
Этот процесс требует затраты определенной энергии Ек, которая является истинной энергией активации гетерогенно-каталитической реакции.
3) реакция в адсорбированном состоянии с образованием адсорбированных конечных продуктов —
(АВК) * → (продукты) К
4) десорбция продуктов реакции с регенерацией катализатора
(продукты) К→ продукты + К
График процесса имеет вид:

 Uпот (АВ) *
Uпот (АВ) *





 Еа
Еа
 ΔНдес
ΔНдес

 ΔНадс продукты
ΔНадс продукты

 А+В АВК (продукты) К
А+В АВК (продукты) К

Как видно из рисунка ΔЕк есть энтальпия адсорбции активного комплекса на катализаторе.
Теории гетерогенного катализа
В настоящее время предложено несколько приближенных теорий, в которых проблема гетерогенного катализа рассматривается на основе различных упрощающих предположений. Согласно современным взглядам, реагирующие вещества образуют с катализатором поверхностные промежуточные соединения. Различия между теориями заключается, в основном, во взглядах на природу поверхностных соединений и на природу активных мест поверхности катализатора. Все теории обычно признают существование активных центров на поверхности катализатора. Изучение адсорбции показало, что поверхность адсорбента неоднородна и различные ее участки обладают разным адсорбционным потенциалом.
Рассмотрим 2 теории гетерогенного катализа:
· Мультиплетная теория Баландина.
В этой теории предполагается, что в образовании поверхностного соединения участвуют группы активных атомов поверхности – мультиплеты, обладающими определенными геометрическими и энергетическими свойствами. В мультиплетной теории рассматриваются принципы геометрического и энергетического соответствия.
Согласно принципу геометрического соответствия, твердое тело может быть гетерогенным катализатором данной реакции, если расположение активных мест на его поверхности находится в геометрическом соответствии с расположением атомов в молекулах реагирующих веществ. Расстояние между атомами в мультиплете должно соответствовать расстоянию между атомами в реагирующих молекулах.
Принцип энергетического соответствия утверждает, что должно быть также определенное соответствие между энергиями связей атомов в молекулах реагирующих веществ и в мультиплетном комплексе, для того чтобы данное твердое тело могло быть катализатором данной реакции.
· Теория активных ансамблей Кобозева.
Предполагается, что активными центрами служат атомы, беспорядочно расположенные на поверхности кристаллического тела. Поверхность твердого кристаллического тела при этом выполняет функцию как бы инертной подкладки. Для каждого данного процесса активным центром является ансамбль из определенного числа атомов нанесенного катализатора. Теория применима в тех случаях, когда на поверхность носителя – твердого тела нанесено очень небольшое число атомов металла, обычно меньше 0,01 от всей поверхности.
Рассмотрим строение поверхности адсорбционного катализатора, когда на нее нанесено небольшое количество металла. Согласно современным взглядам, твердое кристаллическое тело состоит из большого числа микроскопических участков – блоков или областей миграции. Эти участки разделены геометрическими и энергетическими барьерами. При нанесении на твердое тело атомов металла в каждую такую область миграции попадает несколько атомов металла. Область миграции вместе с попавшими в нее атомами металла называется ансамблем. В разных областях миграции может находиться разное число атомов металла, но каталитической активностью обладают только ансамбли с определенным числом атомов металла в внутри области миграции. Такие ансамбли называются активными.
Кинетическая уравнение реакции третьего порядка

Из уравнений (4) и (5) видно, что критериями первого порядка реакции по реагенту А является линейная зависимость ln [ A ] – t или ln  – t
– t
В тоже время по тангенсам углов наклона линейных зависимостей можно определить константы скорости.
Другой тест правильности выбранного первого порядка является постоянство константы скорости реакции, вытекающее из уравнения (5)

Размерность константы скорости первого порядка dim < k >= [1/ c ], [1/мин] или соответственно с -1 , мин -1
Третий тест основан на концентрационной зависимости времени полупревращения. Условие полупревращения [ A ] = 0,5[ A ]0 , тогда в соответствии с уравнением (5)

Можно видеть, что критерием первого порядка реакции является независимость времени полупревращения t ½ от начальной концентрации реагента [ A ]0.
Примерами подобных реакций первого порядка являются реакции изомеризации, а также реакции разложения некоторых сложных молекул в газовой фазе.


и в жидкой фазе, например, гидролиз трет-бутилбромида.

Для реакции A + B → C + D можно записать уравнение скорости

Обозначим [ A ]0 и [ B ]0 – начальными концентрациями реагентов А и В, а Х – количество прореагировавших А и В, тогда уравнение (1) приобретет вид


Разделяя переменные, имеем

Проинтегрируем левую часть этого уравнения методом неопределенных коэффициентов, для чего представим дробь

в виде суммы дробей



Решая совместно эти уравнения, имеем
 ,
, 
Подставляя значения α и β в уравнение (3) и (4) и интегрируя полученные уравнения в соответствующих пределах



Из уравнения (5) видно, что критерием правильности выбранного второго порядка реакции является линейность зависимости
 от времени.
от времени.
По тангенсу угла наклона этой зависимости можно определить константу скорости реакции. Другим критерием правильности выбранного второго порядка является постоянство значений k , вычисленных в соответствии с уравнением (5):

во всем диапазоне пар значений τ – х.
Размерность константы скорости второго порядка
 или, соответственно, л·моль -1 ·с -1 , л·моль -1 ·мин -1 .
или, соответственно, л·моль -1 ·с -1 , л·моль -1 ·мин -1 .
Если вещества А и В взяты в равных количествах или реакция идет с участием одного вещества, например
то при постоянстве объема удобно использовать в качестве переменной концентрацию одного из исходных веществ
тогда кинетическое уравнение будет иметь вид

Интегрируя это уравнение в соответствующих пределах

приходим к выражению


Из уравнений (7) и (8) следует, что критериями правильности выбранного второго порядка являются линейный характер зависимости 1/[ A ] от t и постоянство значения k , вычисленных для различных пар значений [ A ] – t по формуле

Третий критерий правильности второго порядка основан на определении времени полупревращения t ½ . Так как [ A ] = 0.5[ A ]0, то в соответствии с уравнением (7)
 , откуда
, откуда

Можно видеть, что критерием второго порядка является обратно пропорциональная зависимость между t ½ и начальной концентрацией реагента. В соответствии с выражениями (7) и (8) константы скорости второго порядка можно определить по тангенсу угла наклона зависимостей  или
или  от времени.
от времени.
Имеется множество реакций протекающих по кинетике второго порядка:



и простой случай, соответствующий равенству исходных и текущих концентраций реагентов

Разделяя переменные и интегрируя


Из уравнения (3) видно, что критериями правильности выбранного третьего порядка является линейность зависимостей  или
или  от t , постоянства значения k , вычисленное по формуле
от t , постоянства значения k , вычисленное по формуле

для всех пар значений t и [ A ] и обратно пропорциональная зависимость между временем полупревращения и квадратом начальной концентрации реагента

В соответствии с уравнением (3) константа скорости третьего порядка может быть определена по тангенсу угла наклона зависимости  или от времени.
или от времени.

Интегрирование уравнения (1)

приводит к выражениям
Из уравнений (2) и (3) следует, что критериями нулевого порядка по реагенту А являются линейный характер зависимости [ A ] от t , постоянство k , вычисленного по формуле
 во всем диапазоне пар значений t – A и прямолинейная зависимость между временем полу превращения и начальной концентрацией реагента
во всем диапазоне пар значений t – A и прямолинейная зависимость между временем полу превращения и начальной концентрацией реагента 
Из уравнений (2) и (3) следует, что константа скорости нулевого порядка может быть определена по тангенсу угла наклона зависимостей [ A ]0 – [ A ] или [ A ] от времени
Сложные реакции представляют собой совокупность простых реакций. К сложным реакциям относятся обратимые реакции.




При кинетическом анализе сложных реакций руководствуются принципом независимости простых реакций, согласно которому каждая простая реакция, входящая в сложную ведет себя кинетически так, как если бы она была единственная.

в начальный момент времени концентрация реагента A составляет [ A ]0, а [ B ] = 0, то уравнение этой реакции запишется как
Выражая r через концентрацию [A] имеем
 = k1<[A]0 – X> – k-1X
= k1<[A]0 – X> – k-1X
 = k1<[A]0 – X> – k-1X
= k1<[A]0 – X> – k-1X
где X – количество молей вещества A в единице объеме, которое прореагировало к моменту τ и соответственно количество молей вещества B в единице объеме, которое образовалось к этому моменту. Преобразуя правую часть уравнения (2)
= k1[A]0 – (k-1 + k1)X

В условиях равновесия 

При τ ® ¥ X стремится к своему равновесному значению X ® X ¥ . Тогда



где 
Тогда кинетическое уравнение (3) примет вид

Интегрируя это уравнение в соответствующих пределах



и 


В соответствии с выражениями (6) и (7) кинетические зависимости для A и B будут иметь следующий вид

Пользуясь интегральной формой кинетического уравнения (5) и соотношением  можно на основе кинетических данных определить значения констант скоростей k 1 и k -1
можно на основе кинетических данных определить значения констант скоростей k 1 и k -1
Так, в соответствии с (5)

так как  , то
, то

Подставляя последнее выражение в уравнение (8), имеем


Подставляя выражение (10) в уравнение (8), имеем


Рассмотрим систему параллельных реакций первого порядка

В соответствии с ранее принятыми обозначениями суммарная скорость расходования реагента A выразится уравнением

По форме уравнение (1) подобно кинетическому уравнению необратимой реакции первого порядка, поэтому его интегральная форма имеет вид.

Разрешая уравнение (2) относительно [ A ] имеем


Для определения констант k 1 и k 2 рассмотрим уравнения конкурирующих параллельных реакций.


Поделив почленно, левые и правые части уравнений (5) и (6), имеем уравнение  , интегрирование которого приводит к равенству
, интегрирование которого приводит к равенству

Разделяя, левые и правые части уравнений (5) и (1), (6) и (7), приходим к очевидным равенствам
 и
и 
интегрирование которых дает уравнения:
 и
и 
Подставляя в последние уравнения выражение (4) приходим к равенствам


Уравнения (2) и (8) являются основой для определения абсолютных значений констант скорости конкурирующих реакций k 1 и k 2 . На первом этапе можно определить сумму констант скоростей k 1 + k 2 , пользуясь уравнением (2). Затем на основе линейных зависимостей между XB и X , а также XC и X определяют брутто константы  и
и  , из которых рассчитывают k 1 и k 2 по ранее определенному значению суммарной константы скорости k 1 + k 2 .
, из которых рассчитывают k 1 и k 2 по ранее определенному значению суммарной константы скорости k 1 + k 2 .
Нетрудно показать, что для трех параллельных реакций первого порядка


 ,
,
 ,
,

Этот случай более сложен по сравнению с предыдущим. Рассмотрим систему параллельных реакций


Уравнение скорости расхода A в этой системе реакций с учетом его количества, прореагировавшего к моменту времени t (Х) имеет вид:

или с учетом преобразований

обозначая  , имеем
, имеем

Разделяя переменные, приходим к выражению

Интегрируем левую часть уравнения (3) методом неопределенных коэффициентов, для чего представим левую её часть в виде суммы дробей.

 или
или 
так как  , то
, то  и
и 
Тогда  и
и 
Откуда  и
и  ,
, 
С учетом (4) возвращаемся к уравнению (3)

Интегрирование уравнения (5) приводит к выражению

Откуда 
или 
Возвращаясь к соотношению  преобразуем (6) в равенство
преобразуем (6) в равенство



Рассмотрим систему двух последовательных реакций первого порядка

В силу принципа независимости скорости расходования реагента A выражается уравнением скорости необратимой реакции первого порядка

Решение которого дается в виде
 ,
,  и
и

Уравнение скорости изменения концентрации промежуточных продуктов

Разделим почленно левые и правые части уравнений (3) и (1)

Уравнение (4) имеет признаки однородного уравнения первого порядка. Для его решения вводим обозначение 
Подставляя последнее выражение в уравнение (4), имеем:


Разделяем переменные и интегрируем




и 
тогда 
и 
или 
Так как  , то уравнение (5) можно выразить в форме
, то уравнение (5) можно выразить в форме

В тоже время в соответствии с уравнением (2)

Тогда уравнение (5) можно преобразовать в форму

Выведенные зависимости показывают, что в случае необратимых последовательных реакций уравнение для первого промежуточного продукта связано с характеристиками лишь первых двух стадий, оставаясь одинаковым при любом числе и характеристиках последующих стадий. При этом независимо от начальной концентрации реагента A , значение второй характеристики материального баланса  укладывается на одну кривую, если её изображать как функцию ХА или t .
укладывается на одну кривую, если её изображать как функцию ХА или t .
Используя уравнение (5) можно найти  по экспериментальным данным путем подбора и зная, на основе кинетически исчерпывания A величину k 1 – определить k 2 .
по экспериментальным данным путем подбора и зная, на основе кинетически исчерпывания A величину k 1 – определить k 2 .
Из анализа уравнения (6) следует, что при XA = 0 и XA = 1  , что говорит о наличии максимума
, что говорит о наличии максимума  . Его положение можно найти, приравнивая к нулю соответствующую производную
. Его положение можно найти, приравнивая к нулю соответствующую производную

откуда 
и значение максимума

Из выражений (8) и (9) видно, что положение и величина максимума промежуточного продукта в необратимых реакциях первого порядка зависит только от соотношения констант скоростей первых двух стадий. При этом, чем больше величина , тем ниже максимум и тем больше его положение смещается в сторону более низких степеней превращения (и наоборот). Очевидно, что по экспериментальному положению максимума можно определить по специальным номограммам или по уравнению (8) значение и использовать его в дальнейшем для описания значений концентраций B во времени согласно уравнению (7)
Уравнение образования продукта C :


Максимальная скорость  соответствует точке перегиба на зависимости [ C ] от t и определяется из условия
соответствует точке перегиба на зависимости [ C ] от t и определяется из условия


Легко видеть, что это условие соответствует одновременно условию максимума концентрации B , определяемого уравнениями(8) и (9). Качественно проанализированные зависимости могут быть представлены графически.
Неэлементарные реакции состоят из ряда элементарных стадий, составляющих их механизм. Кинетика таких реакций определяется последовательностью элементарных стадий, их характером (обратимые, необратимые), природой реагентов, интермедиатов и продуктов реакции. При кинетическом анализе неэлементарных реакций возникает задача определения концентраций интермедиатов, играющих ключевую роль в образовании продуктов или расходовании реагентов. В качестве инструмента такого определения используется принцип квазистационарных концентраций Боденштейна – Семенова. Согласно этому принципу скорость изменения концентраций нестабильных интермедиатов пренебрежимо мала по сравнению со скоростью изменения концентраций реагентов и продуктов реакции и её можно считать равной нулю. Применение принципа стационарных концентраций к неэлементарным реакциям, протекающим по сложному механизму, позволяет исключить из кинетического описания процессов неизвестные концентрации интермедиатов и получить одно или некоторый минимум дифференциальных уравнений скорости, выраженных через подлежащие измерению концентрации реагентов и продуктов реакции.
Рассмотрим пример неэлементарной реакции, описываемой стехиометрией

и протекающей через образование интермедиата Q


Скорость реакции можно приравнять к скорости образования продукта B

В соответствии с принципом квазистационарных концентраций

откуда 
Подставляя последнее выражение в уравнение (1) приходим к уравнению скорости реакции

Если экспериментально возможно непосредственно измерить скорость реакции, то обработку кинетических данных можно провести, преобразуя уравнение (3) как:

Последнее уравнение приводится к виду

Обрабатывая зависимость (4) в координатах  по ординате находят k 1 , а по тангенсу угла наклона
по ординате находят k 1 , а по тангенсу угла наклона  . Полученных констант достаточно для кинетического описания реакции, так как, разделив числитель и знаменатель уравнения (3) на k 2 , приходят к уравнению
. Полученных констант достаточно для кинетического описания реакции, так как, разделив числитель и знаменатель уравнения (3) на k 2 , приходят к уравнению
http://kursak.net/fizicheskaya-ximiya-ximicheskaya-kinetika-i-kataliz/
http://www.trotted.narod.ru/physchem/26.htm