Молекулярность и порядок химической реакции.
Химические реакции разделяются по признаку их молекулярности или по признаку порядка химической реакции.
Молекулярность химической реакции равна числу молекул (или других частиц), одновременным воздействием между которыми осуществляется элементарный акт химического превращения.
В зависимости от числа таких частиц различают моно (одно-) молекулярные, би (двух-) молекулярные и три (трех-) молекулярные реакции.
Мономолекулярные реакции — реакции разложения молекул:
Для нее закон Гульдберга — Вааге дает:
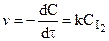 .
.
К бимолекулярным реакциям относятся взаимодействия двух одинаковых или различных молекул:
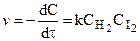 .
.
К тримолекулярным относятся реакции вида:
для которых справедливы соотношения:
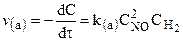 ,
,
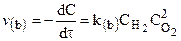 .
.
Молекулярность реакции — понятие теоретическое. Для того, чтобы знать молекулярность, нужно представлять, как именно протекает данная реакция, через взаимодействия каких молекул, через какие стадии.
В противоположность молекулярности порядок реакции — экспериментальная величина. Он связан с опытной зависимостью скорости данной реакции от концентрации исходных продуктов.
Порядок реакции равен сумме показателей степеней концентраций в уравнении, выражающем зависимость скорости реакции от концентрации и устанавливаемом экспериментально.
Для простых реакций, протекающих в полном соответствии с их стехиометрическим уравнением, порядок и молекулярность численно совпадают.
Причины несовпадения порядка реакции и ее молекулярности таковы.
А. Один из реагентов находится в большом избытке. Тогда в ходе реакции его концентрация изменяется незначительно и в уравнении закона Гульдберга — Вааге может быть принята постоянной.
В. Если данная реакция гетерогенная, то в зависимости от условий ее протекания порядок химической реакции изменяется.
С. Порядок каталитических реакций также может отличаться от молекулярности. Причина — сложный механизм таких реакций, не отражаемый стехиометрическим уравнением.
D. Наконец, для сложной реакции, протекающей в несколько стадий, характерно то, что основное влияние на скорость реакции может оказать какая-либо промежуточная стадия, которая и определит, в конечном итоге, порядок всей реакции.
Например, порядок реакции разложения пентаоксида азота
казалось бы, должен быть равен двум.
Исследования показали, что реакция протекает в несколько стадий (результаты анализа химического состава газовой фазы):
причем стадия (1) является наиболее медленной, а поэтому и определяющей скорость всего процесса. Поэтому и суммарная реакция — бимолекулярная реакция первого порядка.
По признаку “порядок химической реакции” различают реакции нулевого, первого, второго и третьего порядков.
Если реакция протекает по нулевому порядку, то
 ,
,
-dC = kd 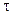
и после интегрирования в интервале от С0 до С за время от 0 до :
С — С0 = -k 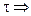 С = С0 — k . (10.6)
С = С0 — k . (10.6)
Следовательно, в реакциях нулевого порядка концентрация линейно уменьшается со временем. Уравнение (10.6) — кинетическое уравнение реакции нулевого порядка.
Период полураспада (полупревращения) 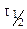 , равный времени, за которое концентрация исходного продукта уменьшается в два раза (С =
, равный времени, за которое концентрация исходного продукта уменьшается в два раза (С = 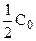 ) составит:
) составит:
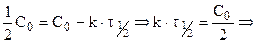
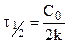 . (10.7)
. (10.7)
Константа скорости может быть найдена из (10.6):
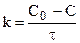 . (10.8)
. (10.8)
Выражение для скорости реакции первого порядка:
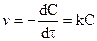 .
.
После разделения переменных и интегрирования от 0 до при изменении концентрации от С0 до С:
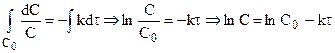 , (10.9)
, (10.9)
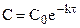 , (10.10)
, (10.10)
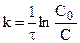 . (10.11)
. (10.11)
Подставляя 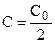 , уравнение для расчета периода полураспада:
, уравнение для расчета периода полураспада:
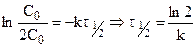 . (10.12)
. (10.12)
Таким образом, для реакции первого порядка выполняются следующие закономерности: зависимость концентрации от времени экспоненциальная; зависимость логарифма концентрации от времени линейная; период полураспада не зависит от начальной концентрации.
При равенстве концентраций исходных продуктов реакции кинетические уравнения реакций второго и третьего порядков соответственно запишутся:
 и
и 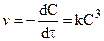 .
.
После разделения переменных:
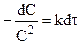 и
и 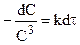 ,
,
и после интегрирования:
 и
и 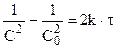 . (10.13)
. (10.13)
Из (10.13) для реакции второго порядка:
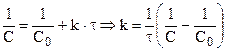 . (10.14)
. (10.14)
Полагая, что для 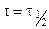 :
:
 . (10.15)
. (10.15)
Для реакции третьего порядка:
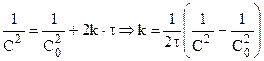 , (10.16)
, (10.16)
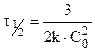 . (10.17)
. (10.17)
Таким образом для реакций второго порядка наблюдается линейная зависимость 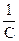 от времени (10.14), а период полураспада обратно пропорционален начальной концентрации. Для реакций третьего порядка наблюдается линейная зависимость от времени величины, обратной квадрату концентрации исходного продукта реакции. Период полураспада таких реакций обратно пропорционален квадрату начальной концентрации.
от времени (10.14), а период полураспада обратно пропорционален начальной концентрации. Для реакций третьего порядка наблюдается линейная зависимость от времени величины, обратной квадрату концентрации исходного продукта реакции. Период полураспада таких реакций обратно пропорционален квадрату начальной концентрации.
При интегрировании кинетических уравнений удобно обозначать концентрации реагентов с помощью изменения концентрации x одного из них в момент времени . Тогда x = C0 — C и уравнения (10.10 и (10.11) примут вид:
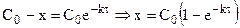 (10.18)
(10.18)
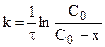 . (10.19)
. (10.19)
Эти уравнения можно получить, составив кинетическое уравнение вида:

и проинтегрировав его в интервале от 0 до при изменении x от 0 до x.
Для реакции второго порядка кинетическое уравнение выглядит:
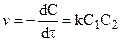 ,
,
если 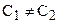 .
.
Тогда с учетом 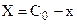 :
:
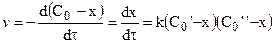 .
.
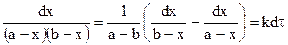 ,
,
Далее после интегрирования:

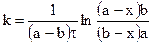 . (10.20)
. (10.20)
Для реакции третьего порядка, ограничиваясь случаем. когда две начальные концентрации одинаковы и равны а (С0’ = C0’’ = a), а третья начальная равна b (С0’’’ = b), уравнение для расчета константы скорости примет вид:
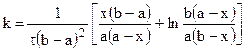 . (10.21)
. (10.21)
Дата добавления: 2015-05-21 ; просмотров: 16560 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Д.Г.НАРЫШКИН
КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИЙ
Возможности компьютерной математики
при исследовании поведения химических систем во времени
МОСКОВСКИЙ ЭНЕРГЕТИЧЕСКИЙ ИНСТИТУТТУ)
ВВЕДЕНИЕ 3
1. ОБЩИЕ ЗАКОНОМЕРНОСТИ ХИМИЧЕСКОЙ КИНЕТИКИ
1.1. Скорость реакции
1.2. Влияние концентрации на скорость реакции
1.3. Молекулярность и порядок реакции
1.4. Прямая и обратная задача химической кинетики
1.5. Реакция первого порядка
1.6. Реакции второго порядка
1.7. Реакции других порядков
1.8. Методы определения порядка реакции
2. Сложные реакции
2.1. Кинетика обратимых реакций
2.2 Параллельные реакции
2.3 Последовательные реакции
2.4 Метод квазистационарных концентраций
3. ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА СКОРОСТЬ ХИМИЧЕСКИХ РЕАКЦИЙ
3.1.Уравнение Аррениуса
3.2. Связь энергии активации с тепловым эффектом реакции
3.3. Связь между скоростью реакции и равновесием
4. КИНЕТИКА ГЕТЕРОГЕННЫХ РЕАКЦИЙ
4.1. Общие понятия
4.2. Макрокинетика. Внешнедиффузионная область
4.3. Макрокинетика. Внутридиффузионное торможение
5. КИНЕТИКА РЕАКЦИЙ В ОТКРЫТЫХ СИСТЕМА
5.1. Химические реакторы.
5.2. Реакторы идеального смешения.
5.3. Реакторы идеального вытеснения.
5.4. Обратимые химические реакции в реакторах
в реакторах смешения и вытеснения.
7. Заключение
8. Рекомендуемая литература
ВВЕДЕНИЕ
Термодинамический метод изучения химических реакций позволяет сделать вывод о принципиальной возможности исследуемого процесса в тех или иных условиях и о глубине его протекания.
При постоянстве давления и температуры самопроизвольное протекание процесса возможно только в направлении уменьшения энергии Гиббса.
Условие 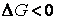 определяет принципиальную возможность проведения процесса в заданных условиях из начального состояния в конечное, но не позволяет оценить скорость такого перехода.
определяет принципиальную возможность проведения процесса в заданных условиях из начального состояния в конечное, но не позволяет оценить скорость такого перехода.
Это обстоятельство связано с тем, что 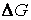 реакции не зависит от пути (механизма) процесса, а определяется только начальным и конечным состоянием системы.
реакции не зависит от пути (механизма) процесса, а определяется только начальным и конечным состоянием системы.
Однако химические реакции могут протекать с самыми различными скоростями – от взрывных до очень медленных, протекающих в течение многих месяцев и лет. Даже одна и та же реакция, протекающая на различных катализаторах, может иметь скорости, различающиеся во много раз.
В некоторых случаях необходимо увеличить скорость реакции, в других наоборот, уменьшить. Таких примеров можно привести множество.
Поэтому изучение скорости протекания химических процессов (а это и составляет задачу кинетики) чрезвычайно важно.
Для рационального проведения химических реакций необходимо уметь управлять ими, знать зависимости скорости от различных параметров.
По кинетике издано очень много учебной и методической литературы. Но все эти книги и учебные пособия написаны так, что хочется спросить: «Какое, милые, тысячелетье на дворе?»
Современные системы компьютерной математики позволяют дать быстрый, и что, пожалуй, главное, наглядный прогноз относительно поведения химической системы во времени.
Однако в русскоязычной учебной литературе по кинетике химических реакций подход, в котором используются средства символьной математики в совокупности со средствами решения систем дифференциальных уравнений, представляемые математическим пакетом Mathcad , практически отсутствует.
Поэтому, отвечая на естественный вопрос – чем предлагаемое учебное пособие отличается от множества других, можно ответить: настоящее пособие имеет цель продемонстрировать эффективность применения математического пакета Mathcad для решения задач химической кинетики.
Специальные химические дисциплины, такие как термодинамика и кинетика, достаточно математизированы, и часто решение химической задачи вызывает у студентов значительные трудности, связанные с математикой – довольно часто это приводит к тому, что приходится сознательно упрощать задачу.
Пособие иллюстрирует богатейшие возможности, которые открывает применение компьютерной математики перед исследователем для анализа поведения химических систем во времени.
В этом отношении математические пакеты становятся практически незаменимыми элементами обучения, позволяющими сделать акцент на содержательном анализе полученных результатов.
Знаком >>>>>> в тексте пособия отмечен переход к Mathcad документу для интерактивного расчета.
1. ОБЩИЕ ЗАКОНОМЕРНОСТИ ХИМИЧЕСКОЙ КИНЕТИКИ
1.1. Скорость реакции
Х имическая кинетика – наука о скоростях и закономерно-стях протекания химических процессов во времени.
Химическая кинетика изучает механизм протекания процесса, т.е. те промежуточные стадии, состоящие из элементарных актов, через которые система переходит из начального состояния в конечное.
Химическая кинетика изучает скорости этих стадий и факторы, влияющие на их скорость.
Уравнение химической реакции показывает начальное состояние системы (исходные вещества) и её конечное состояние (продукты реакции), но не отражает механизма процесса. Однако путь перехода системы из начального в конечное состояние может быть достаточно сложным и «извилистым».
Так, например, реакция
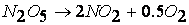
протекает по следующему механизму:
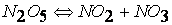
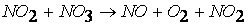
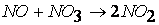
Изучить кинетику реакции – значит показать, как реально протекает исследуемая реакция, её механизм, получить зависимость, связывающую скорость реакции с факторами, влияющими на неё.
Различают два типа химических реакций: гомогенные и гетерогенные.
К гомогенным относят реакции, у которых и исходные вещества и продукты реакции находятся в одной фазе. Взаимодействие веществ в таких реакциях происходит по всему объёму.
К гетерогенным реакциям относят реакции, протекающие на границе раздела фаз.
Пусть протекает реакция
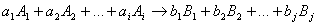 (1.1)
(1.1)
где a 1 , a 2 , ai , b 1 , b 2 , bj – стехиометрические коэффициенты.
Скорость реакции по i –му веществу в гомогенной системе определяется как количество i -го вещества, образующееся (или реагирующее) в единице реакционного объёма в единицу времени:
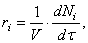 (1.2)
(1.2)
где V – объём реакционной зоны, Ni – количество i –го вещества.
Если реакция протекает изохорически, т.е. объём во время реакции не меняется, то, поскольку концентрация и объём связаны соотношением
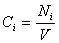 ,
,
скорость реакции можно определить как изменение концентрации вещества во времени
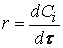
Ранее мы определили скорость химической реакции как изменение числа молей реагирующих веществ в единицу времени в единице объема, т. е.
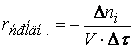
где 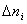 — изменение числа молей одного из исходных веществ за время
— изменение числа молей одного из исходных веществ за время 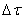 .
.
Таким образом определяется средняя скорость реакции для заданного интервала времени.
Если объем в процессе реакции постоянен, то
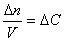
где 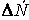 — изменение концентрации.
— изменение концентрации.
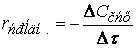 или
или 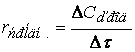
(скорость всегда положительна, а 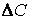 может быть больше или меньше нуля в зависимости от того, изменение концентрации исходного вещества или продукта реакции мы рассматриваем).
может быть больше или меньше нуля в зависимости от того, изменение концентрации исходного вещества или продукта реакции мы рассматриваем).
Если интервал времени 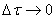 , то мы получим истинную скорость реакции r в данный момент времени, т. е.
, то мы получим истинную скорость реакции r в данный момент времени, т. е.
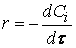
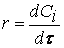 (1.3)
(1.3)
Размерность скорости: моль/(л·с).
Не только знак, но и абсолютное значение скорости зависит от того, по какому из участников реакции она измерена.
Так, например, при протекании реакции
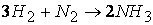
скорость, с которой уменьшается концентрация водорода во время процесса, в три раза больше скорости убывания концентрации азота и в полтора раза выше скорости возрастания концентрации аммиака.
Следовательно, для реакции
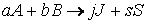
скорости по компонентам реакции будут связаны соотношением:
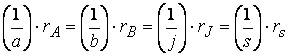
Экспериментально установлено, что скорость реакции зависит от природы реагирующих веществ, их концентрации (или давления), температуры, т.е.
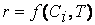
Раскрытие этой зависимости и составляет одну из задач кинетики.
1.2. Влияние концентрации на скорость реакции
Подход к выяснению зависимости скорости реакции от концентрации реагирующих веществ можно иллюстрировать следующим положением теории вероятностей: вероятность одновременного осуществления независимых событий равна произведению вероятностей каждого из них.
Для того чтобы произошло химическое взаимодействие, например, реакция
необходимо, но не достаточно, столкновение реагирующих молекул А и В, т.е. одновременное нахождение их в определённой точке реакционного пространства.
Вероятность ω нахождения молекулы для каждого из веществ прямо пропорциональна количеству молекул в единице объёма, т.е. его концентрации:
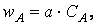 ,
, 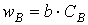 .
.
Тогда вероятность того, что обе молекулы будут одновременно находиться в одной точке пространства, т.е. что они столкнутся, равна
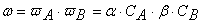
Но не все столкновения приведут к реакции, а лишь их некоторая доля α , величина которой зависит от температуры и природы веществ, поэтому скорость реакции
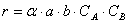
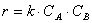
Постоянную k , не зависящую от концентрации и зави сящую только от температуры и природы реагирующих веществ, называют константой скорости реакции.
Численное значе ние k выражает скорость реакции, когда концентрации реагирующих веществ равны 1 моль/л.
Пусть протекает химическая реакция:
aA + bB + … → продукты.
Тогда зависимость скорости реакции от концентрации можно выразить соотношением
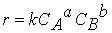 (1.4)
(1.4)
Полученное выражение называют законом действия масс.
1.3. Молекулярность и порядок реакции
Число молекул, вступающих в реакцию, определяют молекулярность реакции.
Так, если в реакцию вступает одна молекула, то такая реакция называется молекулярной реакцией. Если в реакции участвуют две молекулы (безразлично, одинаковые или нет), то такая реакция называется бимолекулярной. Встречаются также тримолекулярные реакции.
Реакции более высокой степени молекулярности крайне редки из–за малой вероятности одновременного столкновения большого числа молекул.
Поэтому большинство реакций протекают в несколько элементарных, простых стадий, в которых участвует небольшое число молекул.
Так, например, рассмотренная выше реакция
протекает по следующему механизму:
вторая стадия (медленная)
Определить такие стадии – значит определить механизм, или путь реакции.
Скорость всей реакции определяется скоростью её наиболее медленной стадии, которая и определяет механизм.
Поэтому закон действующих масс справедлив только для таких элементарных стадий.
Молекулярность реакции легко определить в случае простых реакций, протекающих в одну стадию. В большинстве же случаев довольно трудно найти молекулярность реакции.
Поэтому вводится понятие порядка реакции, который можно найти из кинетических уравнений, полученных экспериментально.
Порядок реакции по данному веществу равен степени, в которой концентрация данного вещества входит в уравнение скорости реакции.
Сумма показателей степеней, в которых концентрация всех исходных веществ входит уравнение скорости реакции, равна порядку реакции в целом. Порядок химической реакции по веществу совпадает со стехиометрическим коэффициентом реакции лишь в очень простых реакциях, например в реакции синтеза йодистого водорода:
Порядок этой реакции по водороду (первый) и йоду (первый) равны стехиометрическими коэффициентами, а общий порядок реакции (второй) равен сумме стехиометрических коэффициентов в уравнении скорости реакции
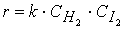
В подавляющем большинстве случаев порядок реакции по веществу отличается от стехиометрических коэффициентов уравнения реакции для этого вещества.
Соответственно общий порядок реакции обычно не равен сумме стехиометрических коэффициентов уравнения реакции.
при температурах, меньших 298К, протекает по следующему механизму:
первая стадия процесса: NO 2 + NO 2 ® NO 3 + NO
вторая стадия процесса: NO 3 + CO ® CO 2 + NO 2,
причем лимитирующей, т.е. скорость определяющей стадией является первая стадия процесса:
Тогда, согласно первому постулату химической кинетики, который утверждает, что скорость всей реакции равна скорости его самой медленной стадии, можно записать:
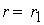 ,
,
где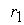 — скорость первой стадии процесса.
— скорость первой стадии процесса.
Согласно второму постулату химической кинетики, который утверждает, что скорость элементарной (одностадийной) реакции пропорциональна концентрации реагирующих веществ в степенях, равных стехиометрическим коэффициентам, получим зависимость скорости реакции
от концентрации реагирующих веществ:
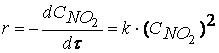
Обратите внимание, что скорость реакции
не зависит от концентрации оксида углерода CO .
Уравнение, выражающее зависимость скорости реакции от концентрации каждого вещества, называют кинетическим уравнением реакции в дифференциальной форме.
К сожалению, кинетическое уравнение реакции может быть получено только при её экспериментальном изучении и не может быть выведено из стехиометрического уравнения.
1.4. Прямая и обратная задача химической кинетики
Определение на основании экспериментальных данных о зависимости концентраций от времени проведения процесса параметров кинетического уравнения – порядка реакции и значения константы скорости – составляет так называемую обратную задачу химической кинетики.
Знание кинетического уравнения реакции в дифференциальной форме позволяет определить время достижения некоторой заданной концентрации реагирующего вещества (или продукта реакции).
Пусть, например, протекает реакция
aA + bB + … → продукты,
кинетическое уравнение которой:
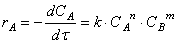
Тогда время достижения некоторой концентрации вещества А можно определить, интегрируя кинетическое уравнение реакции в дифференциальной форме:
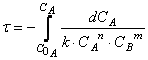
Решая дифференциальное уравнение
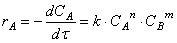
можно получить зависимость концентрации реагирующего вещества (или продукта реакции) от времени проведения процесса – так называемых кинетических кривых.
Определение – на основании феноменологической модели процесса – концентраций реагентов от времени проведения реакции составляет прямую задачу химической кинетики.
Отметим сразу, что аналитически не всегда удаётся решить дифференциальное уравнение, особенно в случае сложной кинетики.
В этом случае прибегают к численным методам решения и использование компьютерной математики. В частности, применение математических пакетов, например, таких, как Mathcad , становится незаменимым инструментом в исследовательской практике и в процессе обучения.
1.5. Реакция первого порядка
Реакция первого порядка может быть записана в общем виде:
Примером такой реакции может служить реакция разложения диметилового эфира:
Кинетическое уравнение реакции первого порядка можно представить дифференциальным уравнением
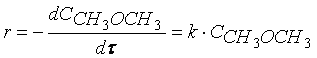 (1.5)
(1.5)
Тогда время t достижения некоторой концентрации диметилового эфира CH 3 OCH 3 можно определить, интегрируя соотношение (1.5):
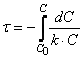 ,
,
где С и C 0 – концентрация CH 3 OCH 3 в момент времени t и t =0.
Интегрирование приводит к выражению
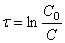 (1.6)
(1.6)  И тогда зависимость концентрации исходного вещества CH 3 OCH 3 от времени проведения процесса:
И тогда зависимость концентрации исходного вещества CH 3 OCH 3 от времени проведения процесса:
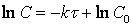 . (1.7)
. (1.7)
Из (1.7) следует, что концентрация исходного вещества со временем изменяется по экспоненциальному закону:
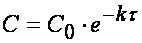
Проиллюстрируем изменение концентрации в зависимости от времени на примере реакции первого порядка
с начальной концентрацией  моль/л и константой скорости при некоторой температуре k=0.05 1/c
моль/л и константой скорости при некоторой температуре k=0.05 1/c
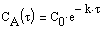


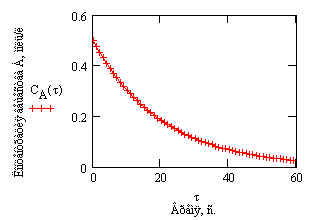
Рис.1. Зависимость концентрации
от времени в реакции первого порядка .
и, в логарифмических координатах, согласно зависимости
Кинетическое уравнение порядок и молекулярность реакции
MedBioGoogle
Навигация
Основные понятия химической кинетики. Химическая реакция.
Химической реакцией можно считать любое изменение вещества, при котором образуется или разрывается химическая связь между атомами. Химическая реакция характеризуется механизмом ее протекания и глубиной протекания.
Химические реакции, как правило, не происходят путем непосредственного взаимодействия исходных молекул с прямым переходом их в молекулы продуктов реакции. В большинстве случаев реакции протекают в несколько стадий. Например, окисление ионов двухвалентного железа в кислом растворе молекулярным кислородом состоит из ряда стадий:
Fe 2+ + HO2 × ® Fe 3+ + HO2 — Общее стехиометрическое уравнение
H2O2 + Fe 2+ ® Fe 3+ + OH — + OH × 4Fe 2+ + 4H + + O2 ® 4Fe 3+ + 2H2O
Fe 2+ + OH × ® Fe 3+ + OH —
Этот сложный путь оказывается тем не менее неизмеримо более выгодным,. Так как на одном из семи этапов не требуется встречи более чем двух частиц, и ни на одном из этапов не требуется соударение одноименно заряженных частиц. Совокупность стадий, из которых складывается химическая реакция, носит название механизма химической реакции.
Исходные, конечные и промежуточные вещества.
Вещества, вступающие в процесс химического превращения, называются исходными веществами. Вещества, образующиеся в процессе химического превращения и не претерпевающие в ходе этого процесса дальнейших химических изменений, называются продуктами реакции. Вещества, образующиеся в одних стадиях процесса химического превращения и расходующегося в других стадиях этого же процесса, называют промежуточными веществами.
Глубина превращения реакции.
Характеризует степень превращения исходных веществ в конечные продукты реакции. Проведенные измерения привели химиков к убеждению, что, во-первых, все мыслимые реакции в какой-то степени происходят, и во вторых, не существует реакций, идущих полностью до конца. Однако, часто удобнее говорить, что одни реакции совсем не идут, другие идут в ограниченной степени, а третьи потребляют исходные вещества полностью.
Гомогенные и гетерогенные реакции.
Химическая реакция, протекающая в пределах одной фазы, называется гомогенной химической реакцией.
Химическая реакция, протекающая на границах раздела фаз, называется гетерогенной химической реакцией.
Примером гомогенных реакции может служить любая реакция в растворе. Примером гетерогенной реакции может служить любая из реакций, идущих не поверхности твердого катализатора (гетерогенная каталитическая реакция).
Скорость химической реакции.
Важнейшей количественной характеристикой процесса химического превращения является скорость процесса. Понятие скорости характеризует количество вещества, вступающего в реакцию в единицу времени. Это определение однако, не совсем однозначно, ак как в реакции принимают участие в качестве исходных и промежуточных веществ и в качестве продуктов реакции также несколько веществ. Поэтому строго можно говорить не о скорости химического процесса вообще, а о скорости по некоторому компоненту. Изменение количества этого компонента принято выражать в числе молей N. Таким образом, для гомофазного химического процесса, идущего веществу называется изменение концентрации этого вещества в единицу времени. Пусть концентрация одного из реагирующих веществ 1 момент времени t1 равна С1, а в момент времени t2 равна С2. Тогда средняя скорость реакции ( ` V ) за промежуток времени t2 – t1 равна
Поскольку концентрация вещества (исходного) в процессе реакции убивает, то
C2 C1 и разность C2 – C1 имеет отрицательный знак, т.е. C2 – C1 = — D С. Отсюда
средняя скорость ` V = — ¾¾
Переходя к бесконечно малым изменениям можно отношение — ¾¾ заменить
на — ¾¾ ; В результате производная от концентрации по времени характеризует
dt мгновенную (истинную) скорость химической реакции:
Скорость химической реакции всегда является величиной положительной, отношение же dC/dt может иметь и положительное и отрицательное значение в зависимости от того представляет ли С концентрацию одного из исходных веществ или одного из продуктов реакции. В первом случае dC/dt 0, но так как скорость должна быть положительной, перед производной ставят минус; во втором случае dc/dt > 0 и чтобы скорость реакции имела положительное значение берут производную со знаком плюс. В общем случае кинетическое уравнение имеет вид
V = + ¾¾ . Однако, необходимо учитывать, что измерение по
dt разным веществам скорости не равны, а пропорциональны одна другой. Например, в реакции синтеза аммиака
на каждый исчезающий моль N2 расходуется 3 моля H2 и образуется 2 моля аммиака. Соответствующие скорости реакции соотносятся, как 1: 3: 2.
Скорость реакции имеет размерность [концентр] × [время] -1 . В химической кинетике концентрацию чаще всего выражают в моль/л, а время в секундах . Отсюда скорость химической реакции выражается в моль × л -1 × с -1 .
8.6. Измерение скорости реакции
В химической кинетике широко используется графический метод изображения функциональных зависимостей. Кривая, изображающая зависимость концентрации какого-либо вещества от времени в ходе процесса химического превращения, носит название кинетической кривой. Крутизна кинетической кривой в каждый момент времени характеризует истинную скорость реакции в этот момент времени, т.к. наклон касательной в точке численно равен скорости:V = — dc/dt = tg a
Часто под кинетическими кривыми понимают и другие зависимости изменения какого-либо изменяющегося параметра в ходе химической реакции (изменение рН, электропроводности, показателя преломления, оптической плотности и т.д.). Однако расссчитывать скорость накопления или расходования какого-либо из компонентов реакции, исходя из такой кривой, можно лишь в том случае, если существует и известна однозначная зависимость, связывающая концентрация этого компонента с измеряемым свойством системы.
Порядок реакции и константа скорости реакции
Скорость химической реакции зависит от целого ряда факторов. При заданных внешних условиях (температура, давление, среда в которой проходит процесс скорость является функцией концентрации реагирующих веществ. Зависимость скорости реакции от концентрации реагирующих вещества описывается основным постулатом химической кинетики: скорость реакции в каждый момент времени пропорциональна произведению концентраций реагирующих веществ, имеющихся в данный момент времени, возведенных в некоторые степени. Этот постулат вытекает из физически очевидного предположения о том, что реагируют те молекулы, которые сталкиваются. Известно, что число столкновений зависит от концентрации молекул, поэтому и скорость химической реакции должна определяться теми же факторами. Итак, можно записать для реакции:
А + В ® С + Д можно записать V = k[A] n 1 × B n 1 , где величины n принято называть порядком реакцию по веществу А и В и т.д. Сумму порядков реакции по всем реагирующим веществам называют порядком реакции.
Следует подчеркнуть, что величины n1 и n2 определяются только экспериментальным путем, т.к. для подавляющего большинства реакций порядки реакции по веществу не равнозначны стехиометрическим коэффициентом.
Порядок реакции – величина формальная. Он может быть положительным или отрицательным, целым или дробным, а также нулевым числом. Как было уже указано, порядок реакции определяется опытным путем, и его нельзя предсказать заранее, даже для формально простых реакций. Например, скорость реакции H2 + I2 = 2HI Согласно опытным данным может быть записана следующим образом: V = k[H2][I2], где порядок реакции по водороду и йоду равен единице, а порядок реакции в целом равен 1 + 1 = 2. В этом случае стехиометрическое уравнение правильно изображает элементарный акт реакции.
Однако, в большинстве сложных реакций, протекающих через несколько стадий, когда общее стехиометрическое уравнение не отражает действительного хода реакции показатели степени в уравнении скорости реакции не будут соответствовать стехиометрическим коэффициентам, т.е. закон действия масс для химических реакций выполняется лишь в некоторых частных случаях.
Можно утверждать, что стехиометрическое уравнение реакции устанавливающее пропорции реагентов и продуктов, не определяет механизма протекания этой реакции во времени. Это является причиной, по которой экспериментально найденный порядок не всегда согласуется с уравнением, описывающим реакцию.
Множитель k в уравнении, показывает с какой скоростью идет химический процесс при концентрациях реагирующих веществ, равных
1 моль/л, называют константой скорости химического процесса. Она не зависит от концентрации и характеризует лишь влияние природы реагирующих веществ на скорость их взаимодействия друг, с другом, т.е. константа скорости реакции является мерой реакционной способности молекул.
Размерность констант скорости реакции различного порядка легко получить из выражения для скорости реакции.
Нулевой порядок V = — ¾¾¾ = ko; ko = [c][t] -1
Первый порядок V = — ¾¾¾ = k1c; ko = [t] -1
Второй порядок V = — ¾¾¾ = k2c 2 ; ko = [c -1 ][t] -1
Константы скорости реакций разных порядков имеют разные размерности и, поэтому их сравнение не имеет смысла. Хотя скорость некоторых химических реакций описывается кинетическим уравнением третьего порядка, однако это еще не значит, что они являются тримолекулярными. Все же укажем
Молекулярность реакции определяется числом молекул, одновременно сталкивающихся и приводящим к химическим превращениям. Взаимодействие подобного рода носят название элементарного акта химического превращения, т.е. молекулярность реакции, в отличие от порядка реакции имеет вполне определенный физический смысл. Например, реакция: I2 = 2I – мономолекулярная, т.к. в ее основе лежит распад исходного вещества; реакция H2 + I2 = 2HI – бимолекулярная. Реакция 2NO + H2 = N2O + H2O является примером тримолекулярной реакции. Молекулярность более высокого порядка не встречается, т.к. одновременное столкновение четырех частиц почти невероятно.
В случае сложных реакций, протекающих в несколько стадий, нет смысла говорить о молекулярности реакции в целом, поскольку это понятие имеет отношение применимо только к отдельным стадиям.
Различие между молекулярностью и порядком реакции можно свести к следующему:
1) молекулярность реакции имеет вполне определенный физический смысл, а порядок реакции – величина формальная;
2) порядок реакции может принимать любые значения: целые, дробные и даже отрицательные, численные значения молекулярности ограничены лишь тремя цифрами 1,2,3.
3) Порядок реакции можно использовать для характеристики любых реакций (как сложных, так и простых), понятие “молекулярность” применимо только к элементарным актам химических превращений.
Количественные, соотношения между скоростью реакции и концентрации реагента.
В реакции первого порядка скорость реакции пропорциональна концентрации одного реагента
Для них V = — ¾¾¾ = k c
Для нахождения С в каждый момент времени t это уравнение необходимо проинтегрировать:
— ¾¾¾ = k c ¾¾¾ = k dt или после интегрирования:
ln c = — kt + const. Значение const находим из начального условия, из которых следует, что Со — начальная концентрация в момент времени t = 0. Определим постоянную., как ln Со. Отсюда ln с = — kt + lnCo; ln C — lnCo = — kt; ln C/Co = -kt
или k = 1/t ln ¾¾ ; заменим на десятичные логарифмы, получим k= ¾¾¾ lg ¾ .
Особенность реакции первого порядка заключается в том, что равным промежуткам времени отвечают равные доли Со/С прореагировавшего вещества. Время t, нужное для того, чтобы прореагировала половина Со, называется периодом полупревращения: подставляя в уравнение t1/2 = t и
С = Со/2, получим: t1/2 = ¾¾¾ , т.е. для реакции первого порядка период
K полупревращения t1/2 не зависит от начальной концентрации и служит характеристикой скорости таких реакций.
Реакция второго порядка
Выражение скорости для реакции второго порядка имеет вид: V = -dc/dt= k × c1 × c2 или V = — dc/dt = kc 2 ; Разделяя переменные, получим –dc/c 2 = k dt; После интегрирования получим: 1/c = kt + const; при Co и t = 0 определяем постоянную интегрирования, равную 1/С0. Отсюда 1/С = kt + 1/Co или 1/С – 1/Со = kt;
Период полупревращения для реакции второго порядка не остается постоянным, а обратно пропорционален начальной концентрации:
Уравнение скорости реакции третьего порядка.
Хотя скорость некоторых химических реакций описывается кинетическим уравнением третьего порядка, тем не менее это еще не значит, что они действительно тримолекулярные. Однако оно удобно для вывода кинетического уравнения:
V = — dc/dt = kc 3 ; После интегрирования получим
½ ( ¾¾ × ¾¾¾ ) = kt или ½ ( ¾¾¾ × ¾¾ ) = kt, если а — исходная
С 2 Со 2 ( а-х) 2 а 2
концентрация, (а-х) – количество вещества оставшегося ко времени t. Если реакция протекает по уравнению: 2А + В ® продукты и если [B] не равна [A], то интегрирование выражения для скорости реакции третьего порядка значительно усложняется
Уравнение скорости реакции нулевого порядка.
Если в уравнении скоростми отсутствуют члены, содержащие концентрацию реагирующих веществ, то скорость такой реакции выражается уравнением нулевого порядка.
Реакция нулевого порядка представляет особый интерес в каталитических процессах, когда катализатор, взятый в определенном количестве, может служить фактором, лимитирующим скорость реакции. Такая ситуация встречается в ферментативной кинетике и будет рассмотрена на следующей лекции.
Определение порядка реакции.
Порядок реакции играет существенную роль при изучении и раскрытии механизма реакции. Он в значительной степени зависит от механизма реакции. Порядок реакции определяется опытным путем и его нельзя предсказать заранее, даже для реакции формально похожих.
а) Для определения порядка реакции часто используют способ подстановки. Он заключается в выборе уравнения кинетики реакции (нулевого, первого, второго порядка) при подстановке в которое экспериментальных данных, получается постоянное значение константы скорости реакции.
б) Существует и графический способ определения порядка реакции. Например, для реакции нулевого порядка скорость реакции не зависит от концентрации. Для реакции первого порядка прямолинейной является зависимость ln C от времени. Для реакции второго порядка линейной будет зависимость 1/С от времени.
в) Используется также определение зависимости от концентрации константы полупревращения и т.д.
Теоретические основы химической кинетики.
В основе современной химической кинетики лежат две теории: теория активных соударений и теория активного комплекса.
Теория активных соударений была сформулирована С.Аррениусом в 1889 году. В основе этой теории лежит представление о том, что для протекания химической реакции необходимо соударение между молекулами исходных веществ, а число соударений определяется интенсивностью теплового движения молекул, т.е. зависит от температуры. Но не каждое соударение молекул приводит к химическому превращению: к нему приводит лишь активное соударение. Тот минимальный запас энергии, которым должны обладать молекулы исходных веществ для того, чтобы их соударение было активным, называют энергетическим барьером реакции. Наглядное представление об энергетическом барьере реакции дает графическое изображение энергетики химической реакции.
В качестве абсциссы в этих диаграммах используется так называемая координата реакции. Вообще говоря, она является сложной функцией межатомных расстояний. Но для практических целей и простых молекул можно считать, что она характеризует изменение в межатомных расстояниях, которые происходят при сближении исходных молекул, образующих активированный комплекс, и взаимном удалении продуктов реакции при распаде активированного комплекса. По оси ординат откладывается потенциальная энергия всей системы.
То дополнительное количество энергии, которое нужно добавить к средней энергии молекул исходных веществ, чтобы соударение стало активным, называется энергией активации.
Энергия активации ощутимо влияет на значение константы скорости реакции и ее зависимости от температуры: чем больше Еа, тем меньше константа скорости и тем значительнее влияет на нее изменение температуры. Константа скорости реакции связана с энергией активации сложной зависимостью, описанной уравнением Аррениуса: k = A × l -Ea/RT , где А – так называемое число “число соударений” представляет собой число соударений в одну секунду между одной молекулой вещества А и одной молекулой вещества В, заключенных в объеме в 1см 3 .
Однако, наблюдаемые константы скорости реакции, как правило, гораздо меньше, вычисленных по уравнению. Поэтому уравнение для константы скорости реакции видоизменяют следующим образом: k = P × Z × l -Ea/RT , где Z – теоретическое число столкновений, а Р – так называемый фактор вероятности или стерический, учитывает все влияния, вызывающие отклонения от идеального уравнения. Необходимость ориентации может заметно тормозить даже сравнительно простые реакции. Хорошо изученным примером является реакция: Н2 + I2 ® 2НI
Для того, чтобы простое соударение дало две молекулы йодистого водорода, надо, чтобы ориентация молекулы была сходна с той, которая изображена на рис.
Энергия активации этой реакции невелика, но скорость мала. Это связано с довольно жесткими требованиями, предъявляемыми к ориентации реагирующих молекул. Тогда А = РZ, т.е. А характеризует чсло соударений С благоприятной ориентацией и называется предэкспоненциальным множителем.
Используя уравнение Аррениуса, можно определить энергию активации Еа. Для этого уравнение Аррениуса удобно применять в логарифмической форме:
ln k = lnA — Ea/R × 1/T или
Если построить график lg k от 1/Е, то получим прямую отсекающую на оси ординат отрезок, равный lg А и имеющую тангенс угла, равный – Еа/2,303R, т.е. tg a = — Еа/2,303R, откуда Еа = -2,303 R tg a
Из уравнения видно, что константа скорости реакции k является произведением двух сомножителей. Предэкспоненциальный множитель А практически не зависит от температуры, т.к. последняя не влияет на взаимную ориентацию молекул. Экспоненциальный множитель l -Ea/RT , который характеризует долю активных соударений от общего числа двойных соударений, сильно зависит от температуры. Теория двойных соударений внесла в химическую кинетику новые представления об активных соударениях и об энергии активации, но эта теория не рассматривала механизм самого соударения, что является ее недостатком.
Менее строгую зависимость константы скорости реакции от температуры в отличие от уравнения Аррениуса дает правило Вант-Гоффа, которое носит эмпирический характер. В 1884 г. Вант-Гофф установил, что при повышении температуры на 10 о С скорость большинства реакций увеличиатся в 2-4 раза. Математически эта зависимость выражается соотношением:
¾¾¾¾ = ¾¾¾¾ = g , где VT и kT – скорость и константа реакции при
VT kT температуре Т: g = 2 ¸ 4 — температурный коэффициент скорости реакции . В общем случае, если температура изменилась на D Т = Т2 – Т1, то уравнение принимает вид:
Следует отметить, что правило Вант-Гоффа можно использовать лишь в узких интервалах температур. Дело в том, что с повышением температуры g уменьшается и при очень высоких температурах может стать даже меньше единицы. В случае ферментативных реакций даже интервал в 10 о С может быть достаточно большим. Для того, чтобы не ошибиться рекомендуется при использовании g использовать более узкий интервал (2 о , 3 о , 5 о ) и полученные результаты приводить к величине
8.17. Теория активированного (переходного) комплекса (переходного состояния
Эта теория – простейший и исторический первый вариант статистической теории химических реакций. Она разработана Э.Вагнером, М.Поляни, Г.Эйрнингом и М.Эвансом в 30-ых годах двадцатого века.
8.18. Вывод основного уравнения теории переходного состояния.
В основу теории также пополнено представление о столкновении молекул как непременном условии реакции. Но она рассматривает то, что происходит в момент столкновения. Для реакции А+В = С в соответствии с теорией переходного состояния следует: А + В « х * ® С, где х * — переходной комплекс. Что подразумевается под переходным комплексом. После столкновения молекул А и В начинается перераспределение химических связей и образование переходного комплекса и переходной комплекс представляет такое состояние взаимодействующих молекул, когда старые связи еще не полностью разорвались, а новые еще не полностью сформировались. В результате мы имеем состояние промежуточное между А,В и С. Переходное состояние характеризуется непрерывным изменением расстояний между взаимодействующими атомами. В этом существенное отличие переходного комплекса от обычной молекулы, в которой средние расстояния между атомами не зависят от времени. Переходный комплекс также не следует путать с промежуточными веществами. Он является динамической структурой, для образования которой требуется затрата энергии. Энергия, необходимая для перевода реагирующих молекул в состояние переходного комплекса носит название энергии активации. Так как исходные молекулы еще не распались, то энергия перехода в активированное состояние меньше энергии разрыва связей в молекулах исходных веществ: Еа Едиссоциации. Таким образом, формирование переходного комплекса – процесс энергетически более выгодный, чем полный распад, вступающих в реакцию молекул. С другой стороны, превращение активированного комплекса в продукты реакции всегда является экзотермическим процессом.
1) Основной постулат теории переходного состояния состоит в том, что исходные вещества всегда находятся в равновесии с переходным комплексом.
В этом случае k химического равновесия переходного комплекса равна:
k * x.p. = ¾¾¾¾ и концентрация
Затем ПК распадается необратимо с образованием продукта С. (характеристикой (количественной) распада будет частота распада ПК – Р. Из статистической механики известно, что Р зависит только от температуры. Эта зависимость имеет вид: Р = kТ/h, где – постоянная Больцмана; h – постоянная Планка; Т – абсолютная температура. Значит, для данной температуры число Р одинаково для всех переходных состояний, а скорость любой химической реакции определяется лишь концентрации определяется лишь концентрации ПК:
Концентрация ПК связана с концентрацией реагентов и поэтому, подставив их выражения, получим:
К обычной реакции взаимодействия применим закон действия масс:
V = kv[А][B], символ kv употребляется для константы скорости в отличие от константы Больцмана. Приравняем правые части уравнений и получим:
Из уравнения видно, что при данной температуре константа скорости реакции зависит от константы химического равновесия образования ПК и от частоты распада ПК. Уравнение называется основным уравнением теории переходного состояния.
8.19. Термодинамическая форма основного уравнения теории переходного состояния.
Термодинамические данные позволяют связать kx.p. * со стандартной свободной энергией образования переходного комплекса D G ¹ , называемый также свободной энергией активации: D G’ ¹ = -RT ln k * x.p.
откуда k * x.p. = e — D G/RT
Тогда для константы скорости химической реакции можно записать
Из этого уравнения видно, что скорость реакции определяется свободной энергией активации D G ¹ , а не Еа, как это следует из теории активных соударений Аррениуса.
Поскольку D G ¹ = D Н ¹ — Т D S ¹ , то
k * x.p. = e — D S/R — D H/RT
или по теории переходного состояния kv равна:
kV = ¾¾ × × e — D S/R — D H/RT или
kV = ¾¾ × × e — D S/R × e — D H/RT
т.е. мы получили термодинамическую форму основного уравнения теории переходного состояния.
8.20. Сравнение термодинамической формы основного уравнения теории переходного состояния с уравнением Аррениуса.
При сравнении уравнения для ТПС с уравнением Аррениуса видно, что
¾¾ × × e — D S/R соответствует А в уравнении Аррениуса.
Т.е. А = ¾¾ × × e — D S/R
Значит определив опытным путем А, можно рассчитать энтропию активации D S ¹ , которая дает важную информацию о механизме образования активированного комплекса. Например, чем более жестко связывается фермент с субстратом, т.е. образуется большее число связей, тем больше уменьшается D S ¹ . Величина D Н ¹ практически равна энергии активации и при сравнении с выражением получается, что kT ¹
Поскольку Z – теоретическое число столкновений и kT/h практически постоянны, вероятностный фактор Р будет, очевидно, связан с энтропией активации D S ¹ . Если она велика и положительна, то фактор Р велик, и реакция будет быстрой. Конечно, это вывод. Но если D S ¹ отрицательна и мала, то реакция будет медленной, т.е. чем больше возрастание энтропии, тем больше вероятность переходного состояния.
Поскольку kT/h соответствует Z и равно при 298К 6,3 × 10 -12 сек -1 , то это число равно числу активных комплексов, разлагающихся за одну секунду в одном см 3 или количеству соударений в 1 секунду в 1 см 3 .
Таким образом, скорость реакции, согласно теории переходного состояния, зависит от двух факторов.
1. Энергетический фактор — D Н ¹ , энтальпия активации. Чем больше энтальпия активации, тем меньше скорость реакции.
2. Энтропийный фактор — D S ¹ , энтропия активации. Чем больше энтропия активации, тем больше скорость реакции.
Учет энтропийного фактора для кинетики реакций во многих отношениях оказался плодотворным и впервые позволил установить связь константы скорости со строением молекул реагирующих веществ. При этом теория ПС оперирует, в частности, величинами расстояний между атомами в молекулах, взаимной ориентацией молекул. Т.е. параметрами геометрического характера.
Теория активных соударений позволяет при знании энергии активации рассчитать общее число эффективных соударений и отсюда скорость реакции, не объясняя механизма реакции. В отличие от теории активных соударений теория ПК сопоставляет различные возможные комплексы, выявляет большую или меньшую их достижимость и определяет в результате энергетически наиболее выгодный путь реакции.
Для вычисления скоростей взаимодействие двух атомов две теории дают одинаковые результаты. В случае нелинейных многоатомных молекул теория ПК дает значение скоростей отличных от значений, даваемой теорией соударений. Если известна конфигурация реагирующих молекул и активного комплекса, теория ПК позволяет рассчитать предэкспоненциальный множитель. К сожалению, в большинстве случаев строение активированного комплекса и его свойства неизвестны и это затрудняет расчеты.
Таким образом две теории дополняют друг друга. Теория ПК применяется для вычисления абсолютных скоростей электродных процессов, процессов диффузии и т.д. Теория активных соударений хорошо описывает, как правило, реакции в газовой среде.
http://twt.mpei.ac.ru/TTHB/1/Chem/Kin.html
http://www.sites.google.com/site/1bio1med1/home/immunologia/ekologia/biomembranes-and-cell-architecture/pervoe-nacalo-termodinimiki-/vtoroe-nacalo-termodinamiki-i-ego-primenenie-/trete-nacalo-termodinamiki-rascety-izmenenia-entropii-pri-razlicnyh-processah-/himiceskij-potencial-/himiceskoe-ravnovesie-v-geterogennyh-i-gomogennyh-sistemah-/rastvory-elektrolitov-/osnovnye-ponatia-himiceskoj-kinetiki