Кинетическое уравнение реакции кислотно основного катализа
Тогда уравнение скорости между протонированной формой A и Y выразится как

Уравнение (13) отражает противоборствующее влияние кислотности среды на скорость реакции: 1-й член увеличивается с ростом кислотности, но 2-й уменьшается. Такой характер влияния обусловливает экстремальную зависимость скорости соответствующих реакций от кислотности среды. Подобные зависимости наблюдаются для кислотно-каталитических реакций карбонильных соединений с азотистыми основаниями.
Гетерогенный катализ важнейшая область катализа, имеющая широкое практическое приложение. Достаточно в этом плане отметить такие важные процессы как синтез аммиака



В то же время гетерогенно-каталитические реакции – сложные и многомерные физико-химические системы, представляющие большой научный интерес.
Преимущества гетерогенного катализа перед гомогенным состоят в малом расходе катализатора на единицу количества продукта, снижении или полном устранении токсичных сточных вод и расходов дополнительных реагентов на промывку реакционной массы. Это обусловлено тем, что катализатор и реакционная масса находятся в разных фазах.
По способу осуществления гетерогенно-каталитические процессы делятся на процессы со стационарным (неподвижным) катализатором, где он используется в виде достаточно крупных гранул (0,3 – 1 см) и на процессы с подвижным катализатором (плавающим, диспергированным или псевдоожиженным). В последнем случае катализатор применяют только в диспергированном виде, когда он способен перемешиваться под влиянием потока реакционной массы.
Каталитические процессы осуществляются в газовой или жидкой фазе.
В зависимости от типа механизма реакций, протекающих на гетерогенных катализаторах последние подразделяют на следующие группы:
1. Ионные, под влиянием которых протекают реакции по ионному механизму.
2. Электронные, катализирующие гомолитические реакции
3. Бифункциональные, совмещающие ионный и электронный катализ
К ионным катализаторам относятся следующие:
1. Кислотно-основные катализаторы
В группу этих катализаторов входят
а) оксиды некоторых металлов, например Al 2 O 3 , W 2 O 3 , ThO 2 .
б) нейтральные и кислые соли: Ca 3 ( PO 4 )2, CaHPO 4 , MgHPO 4 , а также природные и синтетические алюмосиликаты ( Al 2 O 3 ) m ( SiO 2 ) n ( H 2 O ) p .
в) Протонные и апротонные кислоты на носителях ( H 3 PO 4 на Al 2 O 3 или кизельгуре, BF 3 на Al 2 O 3 , гетерополикислоты) и ионообменные смолы.
2. Комплексообразующие соли переходных металлов на носителях: Cu 2 Cl 2 , HgCl 2 , PdCl 2 + CuCl 2 , CuC º C С u , NiCl 2 и другие.
Все катализаторы ионного типа являются изоляторами или ионными проводниками электрического тока. Наиболее распространены катализаторы кислотного типа, являющиеся протонными (бренстедовскими) или апротонными (льюисовскими) кислотами.
Ионообменные смолы используют главным образом для катализа жидкофазных реакций, которые и в гомогенных условиях катализируются протонными кислотами (дегидратация, этерификация, алкилирование, конденсация по карбонильной группе) и только до температуры » 150 ° C , выше которой ионообменные смолы склонны к деструкции. Для тех же но газофазных реакций и гидратации олефинов используют фосфорную кислоту на носителях. Оксиды и соли применяются как для перечисленных выше реакций, так и для крекинга углеводородов (алюмосиликат) или алкилирования аминов ( Al 2 O 3 ) Соли обладают мягким действием (например, фосфаты), что обусловило их использование для тех реакций, реагенты или продукты которых чувствительны к влиянию более активных катализаторов.
Гетерогенные катализаторы гомолитических реакций всегда являются электронными проводниками тока или полупроводниками, они включают следующие группы веществ:
1. Перехолные металлы I Б и VIII групп ( Cu , Ag , Fe , Ni , Co , Pt , Pd ) периодической системы Д. И. Менделеева.
2. Оксиды металлов ( MgO , ZnO , CuO , Fe 2 O 3 , Cr 2 O 3 , WO 3 , MoO 3 , V 2 O 5 ) сульфиды ( WS 3 , MoS 3 ) и смеси, содержащие один основной оксид с небольшими добавками других (модифицированные оксидные катализаторы)
3. Сложные оксидные и сульфидные катализаторы с соизмеримым соотношением компонентов, а также соли-полупроводники: хромиты ( CrO × Cr 2 O 3 , ZnO × Cr 2 O 3 ), вольфраматы ( CoO × WO 3 ), молибдаты ( Bi 2 O 3 × MoO 3 , NiS × MoS 3 ) ванадаты и другие.
Между ионными и электронными катализаторами не существует резкой границы, некоторые из них способны ускорять ионные и гомолитические реакции. Из индивидуальных веществ это особенно относится к оксидам, часть из которых катализирует ионные, а остальные – гомолитические реакции. Относительная активность оксидных катализаторов в ионных и гомолитических реакциях может быть проиллюстрирована на примере конкуренции двух реакций – ионной реакции дегидратации этанола и гомолитический реакции – его дегидрирования.
относительная доля продукта дегидратации, %
относительная доля продукта дегидратации, %
Из приведенных данных видно, что некоторые оксиды являются бифункциональными катализаторами. Бифунциональности катализаторов можно также достичь используя смеси оксидов разного типа. Лучшим примером является система ZnO на Al 2 O 3 , успешно применявшаяся для синтеза бутадиена из этанола, который представляет собой совмещенный процесс дегидратации, димеризации и дегидрирования. Другой пример бифункциональных катализаторов – металлы платиновой группы на носителях кислотного типа ( Al 2 O 3, алюмосиликаты), являются катализаторами каталитического риформина в котором протекают гомолитические реакции дегидрирования и ионные реакции изомеризации и расщепления.
Гетерогенные катализаторы должны удовлетворять следующим требованиям:
— высокая каталитическая активность
— достаточно большая селективность (избирательность) в отношении целевой реакции
— простота получения, обеспечивающая воспроизводимость всех свойств катализатора.
— высокая механическая прочность к сжатию, удару и истиранию
— достаточная стабильность всех свойств катализатора на протяжении его службы и способность к их восстановлению при том или ином методе регенерации.
— небольшие экономические затраты на катализатор при производстве единицы продукции.
Обеспечение этих требований достигается главным образом при разработке состава катализатора и способа его получения.
Гетерогенные катализаторы сравнительно редко применяются в индивидуальном виде и часто содержат различные добавки, получившие название модификаторов. Цели их введения разнообразны: повышение активности катализатора (промоторы), избирательности и стабильности работы, улучшение механических или структурных свойств. Фазовые и структурные модификации стабилизируют соответственно активную фазу твердого катализатора или пористую структуру его поверхности. Так, в меди-хромистых катализаторах гидрирования оксид хрома препятствует восстановдению оксида меди с превращением его в неактивную форму. Добавление уже 1% Al 2 O 3 к железному катализатору значительно увеличивает его поверхность, препятствуя спеканию и закрытию пор. Некоторые модификаторы существенно повышают стабильность работы катализатора.
В смешанных катализаторах, где компоненты находятся в соизмеримых количествах, могут образовываться новые более активные соединения, их твердые растворы в основном компоненте или многофазные системы, обладающие специфическим каталитическим действием. При этом свойства смешанного катализатора не являются простой суммой свойств его компонентов. Это приводит к заметному увеличению избирательности, активности и других свойств катализатора.
К числу модификаторов можно отнести и носители (трегеры), особенно часто применяемые в случае дорогостоящих металлических катализаторов ( Pt , Pd , Ni , Co ). Роль последнего состоит в повышении активной поверхности, увеличении термостойкости и механической прочности катализатора и т.д. В качестве последнего используют алюмосиликаты, Al 2 O 3 , Cr 2 O 3 , SiO 2 , активированный уголь, пемзу, кизельгур и другие природные и синтетические материалы.
Как правило, гетерогенно-каталитический процесс протекает через ряд последовательных, а иногда и последовательно параллельных стадий, существенно различающихся по своей природе. К таким процессам относятся:
1. диффузия реагентов из потока к внешней поверхности зерна катализатора.
2. диффузия реагентов к внутренней поверхности зерна катализатора (в поры)
3. адсорбция реагентов на поверхности катализатора
4. собственно химическая реакция
5. десорбция продуктов реакции с поверхности катализатора
6. диффузия продуктов с внутренней поверхности зерна катализатора
7. диффузия продуктов с внешней поверхности зерна катализатора в поток
Любая из этих стадий может оказаться лимитирующей и, следовательно, определять скорость процесса в целом. Таким образом, кинетические закономерности гетерогенно-каталитического процесса могут контролироваться закономерностями диффузии, адсорбции или химической реакции, а в пограничных случаях – их совокупностью. Это определяет специфику кинетики гетерогенного катализа по сравнению с кинетикой гомогенного катализа.
Принято различать кинетически- и диффузионно- контролируемые области протекания гетерогенно-каталитических процессов. В первых из них общая скорость процесса определяется скоростью химической реакции на поверхности, во вторых – диффузией реагентов или продуктов реакции. Кроме того, существуют области, контролируемые сорбцией реагентов или десорбцией продуктов.
Более детально различают следующие 5 основных областей:
1. Внешнедиффузионная область. В этой области скорость в целом определяется скоростью транспорта реагентов из потока к внешней поверхности зерна катализатора (или скоростью диффузии продуктов с внешней поверхности в поток)
2. Внутридиффузионная область. В этом случае скорость процесса лимитируется диффузией реагентов от внешней поверхности зерна катализатора к его внутренней поверхности (или наоборот, диффузией продуктов реакции из пор катализатора к его внешней поверхности)
3. Внешнекинетическая область. В этом случае скорость процесса лимитируется скоростью химической реакции на внешней поверхности зерна катализатора. Это возможно, если скорость химической реакции на внешней поверхности значительно превосходит скорость внутренней диффузии (стадии 2 и 6), но значительно меньше скорости внешней диффузии.
4. Внутрикинетическая область. Скорость процесса определяется скоростью химической реакции, причем последняя протекает на внешней и внутренней поверхности зерна катализатора. Это возможно, когда химическая реакция идет значительно медленнее внешней и внутренней диффузии.
5. Сорбционная область. Здесь скорость процесса определяется адсорбцией реагентов или десорбцией продуктов.
Строгие границы между этими областями отсутствуют, они перекрываются переходными областями, в которых сочетаются закономерности различных областей.
При экспериментальном исследовании и расчетах гетерогенно-каталитических процессов важно знать область, в которой протекает реакция, отчего зависит вид описывающих её кинетических уравнений. Так, при лимитировании скорости процесса химической реакцией на всей поверхности катализатора (внутрикинетическая область) скорость диффузии не играет роли, и результаты процесса не будут зависеть от размера зерен катализатора. Наоборот: во внешне- или внутридиффузионной области размер зерна играет большую роль, так как скорость диффузии на единицу массы катализатора зависит от величины внешней поверхности, которая определяет и диффузию в поры. Таким образом, проводя серию экспериментов с катализатором разного размера зерна и наблюдая за изменением конверсии, можно различить кинетическую и диффузионную области, а также определить размер зерна катализатора, необходимый для достижения кинетической области.
Тестом на различие внешне- и внутридиффузионной областей является зависимость скорости внешней диффузии от линейной скорости газового потока, тогда как внутренняя диффузия не зависит от этого параметра. Линейную скорость потока можно варьировать, меняя объем катализатора, соблюдая при этом постоянство времени контакта.

Легко убедиться, что такое требование можно соблюсти при условии пропорционального изменения объема υ линейной скорости n , связанной с объемной скоростью потока соотношением

Изменение или постоянство степени конверсии в таких опытах свидетельствует о наличии или отсутствии внешнедиффузионного торможения.
При исследовании гетерогенно-каталитических реакций наиболее достоверные данные получаются в проточно-циркуляционных установках, хотя часто используют метод исследования в потоке.
Во избежание нарушений в режиме потока реагентов диаметр каталитической ячейки или трубы должен быть равным не менее 6-7 диаметров зерна катализатора.
Обычно при работе катализаторов происходит постепенное изменение их активности и селективности. Поэтому кинетический эксперимент можно ставить лишь в период их стационарных свойств, который наступает после более или менее длительного холостого пробега и заканчивается при наличии признаков заметной дезактивации катализаторов.
Кинетические эксперименты проводят при варьировании начальных концентраций (парциальных давлений) реагентов и продуктов, а также условного времени контакта.
При кинетическом анализе реакций на поверхности катализатора руководствуются законом действующих поверхностей, согласно которому скорость химической реакции пропорциональна поверхностной концентрации реагентов, участвующих в акте химического взаимодействия.
В случае мономолекулярных реакций, например A ® B , акт химического взаимодействия можем представлять собой превращение вещества, сорбированного на активном центре поверхности, или его взаимодействия с соседним свободным центром поверхности. Эти двум механизмам соответствуют кинетические уравнения

В случае бимолекулярных реакций, например A + Y ® B , возможны также два механизма. Согласно одному из них два реагента адсорбируются на разных активных центрах поверхности и взаимодействуют между собой, если эти центры находятся в достаточной близости друг от друга (механизм Хиншельвуда). Такому механизму соответствует следующее кинетическое уравнение

Согласно другому механизму на поверхности сорбируется один из реагентов, активирования которого достаточно для взаимодействия с налетающей из объема молекулой второго реагента (“ударный” механизм Ридила). Такому механизму соответствует кинетическое уравнение

Неэлементарные реакции состоят из ряда элементарных стадий, составляющих их механизм. Кинетика таких реакций определяется последовательностью элементарных стадий, их характером (обратимые, необратимые), природой реагентов, интермедиатов и продуктов реакции. При кинетическом анализе неэлементарных реакций возникает задача определения концентраций интермедиатов, играющих ключевую роль в образовании продуктов или расходовании реагентов. В качестве инструмента такого определения используется принцип квазистационарных концентраций Боденштейна – Семенова. Согласно этому принципу скорость изменения концентраций нестабильных интермедиатов пренебрежимо мала по сравнению со скоростью изменения концентраций реагентов и продуктов реакции и её можно считать равной нулю. Применение принципа стационарных концентраций к неэлементарным реакциям, протекающим по сложному механизму, позволяет исключить из кинетического описания процессов неизвестные концентрации интермедиатов и получить одно или некоторый минимум дифференциальных уравнений скорости, выраженных через подлежащие измерению концентрации реагентов и продуктов реакции.
Рассмотрим пример неэлементарной реакции, описываемой стехиометрией

и протекающей через образование интермедиата Q


Скорость реакции можно приравнять к скорости образования продукта B

В соответствии с принципом квазистационарных концентраций

Откуда 
Подставляя последнее выражение в уравнение (1) приходим к уравнению скорости реакции

Если экспериментально возможно непосредственно измерить скорость реакции, то обработку кинетических данных можно провести, преобразуя уравнение (3) как:

Последнее уравнение приводится к виду

Обрабатывая зависимость (4) в координатах  по ординате находят k 1 , а по тангенсу угла наклона
по ординате находят k 1 , а по тангенсу угла наклона  . Полученных констант достаточно для кинетического описания реакции, так как, разделив числитель и знаменатель уравнения (3) на k 2 , приходят к уравнению
. Полученных констант достаточно для кинетического описания реакции, так как, разделив числитель и знаменатель уравнения (3) на k 2 , приходят к уравнению

с известными коэффициентами.
Плодотворность метода квазистационарных концентраций может быть проиллюстрирована на примере химических реакций, катализируемых ферментами.

Где Е – активный центр фермента, Х – промежуточное соединение, которое часто называют фермент-субстратным компонентом.
Кинетическая модель этой реакции может быть представлена системой кинетических уравнений


Решение этой системы уравнений в аналитической форме невозможно, поэтому оценка констант уравнений k 1 , k 2 , k 3 и k 4 производится численными методами.
Ферментативные реакции обычно изучают при концентрациях фермента значительно меньших, чем концентрациях субстрата, поэтому можно с высокой степенью приближения принять, что ферментативная реакция протекает стационарно, так что  . Поскольку [ E ]0 = [ E ] + [ X ] где [ E ]0 – начальная концентрация фермента (точнее, его активных центров), уравнение (6) можно представить в виде
. Поскольку [ E ]0 = [ E ] + [ X ] где [ E ]0 – начальная концентрация фермента (точнее, его активных центров), уравнение (6) можно представить в виде
 Разрешая последнее уравнение относительно [ X ], имеем
Разрешая последнее уравнение относительно [ X ], имеем

Вводя обозначение  ,
, 
Приводим уравнение (8) к форме

Подставляя полученное выражение в уравнение (7) с учетом равенства [ E ]0 = [ E ] + [ X ] приходим к уравнению

Уравнение (9) называется уравнением Михаэлиса – Ментен. При низких конверсиях концентраций продукта P в уравнении (9) можно пренебречь и последнее приобретает вид

При [ S ] KS можно пренебречь единицей в знаменателе уравнения (10) и скорость реакции имеет первый порядок по субстрату:

В противоположном случае [ S ] KS можно пренебречь вторым членом в знаменателе уравнения (10) и реакция имитирует нулевой порядок по субстрату

В обоих случаях порядок реакции является первым по концентрации активных центров фермента. Величина u S = k 3 [ E ]0 называется максимальной скоростью ферментативной реакции, а k 3 – числом реакционных центров для прямой реакции. Величина  называется константой Михаэлиса для субстратов. Как видно из уравнения (10) она равна концентрации субстрата, при которой скорость реакции составляет половину максимальной. Значение u S и KS можно найти, преобразуя уравнение (10) в линеаризированную форму
называется константой Михаэлиса для субстратов. Как видно из уравнения (10) она равна концентрации субстрата, при которой скорость реакции составляет половину максимальной. Значение u S и KS можно найти, преобразуя уравнение (10) в линеаризированную форму

Тогда u S определяется по ординате линейной зависимости  от
от  , а KS – по тангенсу угла наклона с учетом найденной величины u S .
, а KS – по тангенсу угла наклона с учетом найденной величины u S .
Параметры u P и KP – могут быть найдены численными методами с учетом известных значений u S и KS путем обработки экспериментальных данных полученных при достаточно высоких конверсиях субстрата.
Механизмы и кинетика кислотно-основного катализа
Механизмы и кинетика кислотно-основного катализа
ГОМОГЕННЫЙ КИСЛОТНО-ОСНОВНЫЙ КАТАЛИЗ
Множество реакций органической химии, в том числе и каталитических, может быть рассмотрена с позиции кислотно-основных взаимодействий. Трактовка механизмов кислотно-основного катализа (как гомогенного, так и гетерогенного) базируется на фундаментальных положениях теории кислот и оснований.
Кислотами или основаниями считаются вещества, проявляющие кислотные или основные свойства. Проявление кислотных или основных свойств зависит от условий среды, в которой находятся эти вещества.
Первый научный подход к определению кислот и оснований был сделан Оствальдом и Аррениусом в 1890 г. после создания последним теории электролитической диссоциации. Это была ионная теория, согласно которой кислота — это водородсодержащее соединение, способное генерировать протон (Н + ), а основание — это вещество, генерирующее гидроксил (ОН — ).
МЕХАНИЗМЫ КИСЛОТНО-ОСНОВНОГО КАТАЛИЗА И ФАКТОРЫ, ОПРЕДЕЛЯЮЩИЕ ЕГО ЭФФЕКТИВНОСТЬ.
Разделяют четыре типа гомогенных катализаторов действующих по механизмам кислотно-основного взаимодействия: 1) нуклеофильные; 2) кислотные; 3)основные; 4)электрофильные.
НУКЛЕОФИЛЬНЫЙ КАТАЛИЗ
Таблица 2.3.
Нуклеофильность ЕNu в реакции нуклеофильного замещения при насыщенном атоме углерода и ее связь с основностью Н и поляризуемостью Р нуклеофила (Nu).
| Nu | Н | Р | ЕNu | Nu | Н | Р | ЕNu |
| F — | 4.9 | -0.149 | -0.23 | HO — | 17.48 | 0.142 | 1.60 |
| H2O | 0 | 0 | 0 | NH3 | 11.22 | 0.184 | 1.84 |
| CH3COO — | 6.46 | — | 0.95 | CN — | 10.88 | 0.383 | 2.01 |
| Cl — | -3.00 | 0.389 | 1.21 | I — | -9.00 | 0.719 | 2.02 |
| C6H5O — | 11.47 | — | 1.46 | S2O3 2- | 3.6 | — | 2.52 |
| Br — | -6.00 | 0.539 | 1.57 | C2H5O — | 18.3 | — | 3.28 |
Из данных таблицы 2.3 видно, что такое сильное основание, как F — в данных условиях является очень слабым нуклеофилом из-за малой поляризуемости, а очень слабое основание I — , наоборот, проявляет высокую нуклеофильную активность.
В тех случаях, когда основность является фактором, определяющим нуклеофильность (aP Nu /КВ H 2 O )
которое еще ранее было предложено Бренстедом в форме:
Корреляционное уравнение Бренстеда хорошо применимо для реакций, контролируемых зарядами взаимодействующих атомов, а также в сериях реакций, в котором атом реакционного центра одинаков и его поляризуемость можно считать постоянной (Р = const), например для нуклеофильных реакций фенолятов, меркаптидов, аминов и др. В соответствии с уравнением (2.36) для серий таких реакций наблюдается линейная зависимость lg(kNu) от lg(КВ Nu ) со значениями b в пределах 0
КИСЛОТНЫЙ КАТАЛИЗ
В органическом синтезе наиболее распространен катализ протонными кислотами. Наиболее используемые из них: серная, соляная, орто-фосфорная, ароматические сульфокислоты (бензолсульфокислоты, толуолсульфокислота), муравьиная и некоторые другие.
Очевидно, что кислотный катализатор способен взаимодействовать с реагентом в том случае, если последний проявляет основные свойства. Катализатор вступает с реагентом в кислотно-основное равновесие как с переносом заряда, так и без:
Н + В ßà АН××××:В ßà А — :××××НВ + ßàА — + НВ +
Протонирование реагента приводит к появлению положительного заряда и сильной поляризации соседних связей, что так или иначе вызывает дефицит электронов на одном из атомов и делает возможным или облегчает дальнейшее превращение. Оно может протекать по двум механизмам: А-1 и А-2.
Механизм А-1 включает стадию мономолекулярного распада протонированной молекулы по s-связи:
RX + H + ßà RXH + ßà R + + XH
Стадия распада лимитирует скорость реакции. Такому механизму благоприятствуют электронодонорные заместители, стабилизирующие карбокатион, который затем по быстрой реакции взаимодействует либо с реагентом-нуклеофилом с образованием продукта замещения:
либо с основанием, отщепляющим протон с образованием ненасыщенного соединения:
Н-С-С + + В à ВН + + С=С
Образующийся на первой стадии карбокатион иногда успевает изомеризоваться в более устойчивый.

По механизму А-1 протекают реакции спиртов, простых и сложных эфиров, азотистых соединений, которые образуют стабильные промежуточные катионы. Например:

Круг реакций, протекающих по механизму А-2 более широкий. Он включает активирование реагента не только за счет протонирования s-связанного заместителя (RXH + ), но и за счет присоединения протона по кратным связям. Дальнейшее превращение протонированного соединения всегда бимолекулярно.
Соединения, протонированные по s-связанному заместителю, подвергаются нуклеофильной атаке вторым реагентом по насыщенному атому углерода. Например:
Для соединений, протонированных по кратным связям, характерны последующее присоединение нуклеофила и отщепление протона:
или последующее присоединение по ароматическим или кратным связям с последующим отщеплением протона:

В реакциях производных карбоновых кислот (RСОX) присоединение нуклеофильного реагента к протонированной карбоксильной группе приводит к последующему отщеплению группы Х и образованию продукта замещения. Например, как при гидролизе сложных эфиров:

Кислотный катализ без полной передачи протона от кислоты к реагенту характерен для слабых оснований-реагентов, слабых кислот-катализаторов и неполярных растворителей. В таких случаях кислотно-основное взаимодействие реагента с катализатором останавливается на стадии образования водородных связей. Например, в отсутствии добавок сильных кислот роль кислотного катализатора при этерификации уксусной кислоты спиртом выполняет вторая молекула уксусной кислоты:
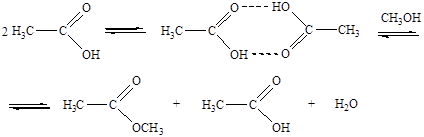
Рисунок 2.3.
Кинетический тест на наличие общего или специфического катализа: сплошные линии — общий катализ; пунктир — специфический катализ; рН1 > рН2.
Общий кислотный катализ
При общем кислотном катализе скорость реакции зависит от концентрации каждой из форм кислотного катализатора, присутствующей в реакционной массе. Общий кислотный катализ проявляется в тех случаях, когда стадия присоединения протона к реагенту является лимитирующей, а дальше идут быстрые реакции превращения активированных молекул. В общем виде механизм реакций можно представить следующей схемой:
медленно: 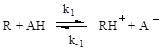
быстро: 
Образовавшийся на первой (лимитирующей) стадии активированный реагент (RH + ) далее быстро превращается в продукт реакции (P) по мономолекулярной реакции или при участии второго реагента (Y). В соответствии с данной схемой скорость реакции лимитируется протолитической реакцией (2.56) и кинетическое уравнение имеет вид:
При этом, если катализатор присутствует в разных формах (AiH), то каждой из них соответствует своя константа скорости (ki), определяемая активностью данной формы катализатора, и, в общем виде, уравнение (2.57) можно представить так:

Рассмотренная схема характерна для реакций, в которых на первой стадии происходит медленное образование С-Н связи. Примером служат кислотно-каталитические реакции олефинов, протекающие с образованием карбокатионов:

Но бывают и другие случаи. Известно множество примеров, когда лимитирующей оказывается стадия передачи протона между атомами катализатора и реагента. Это происходит, когда перенос протона сопровождается синхронным разрывом или образованием новой связи. Этому обычно предшествует быстрая стадия координации кислоты и основания за счет образования водородной связи. Общая схема для таких случаев выглядит так:
Образование водородной связи (быстро):
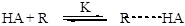

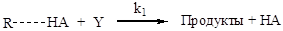
Согласно приведенной схемы (2.60), скорость реакции будет определяться скоростью второй (лимитирующей) стадии, и кинетическое уравнение (например, для бимолекулярного превращения) будет иметь следующий вид:
Но при высоких значениях константы равновесия первой стадии или при высоких концентрациях реагента, что обеспечивает количественное связывание катализатора в активированный комплекс с реагентом (т.е. когда [R×××HA] = [HA]), уравнение принимает более простой вид:
Примерами бимолекулярных превращений активированного комплекса при общем кислотном катализе могут служить реакции этерификации:

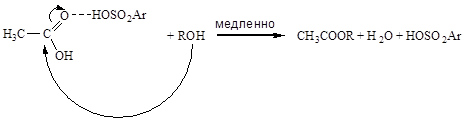
ЭЛЕКТРОФИЛЬНЫЙ КАТАЛИЗ
Электрофильный катализ осуществляется кислотами Льюиса. Электрофильными катализаторами в органических реакциях могут выступать нейтральные молекулы, соли металлов, ионы и некоторые другие соединения, например:
Ионы металлов — Li + , Ag + , Hg + ;
Роль электрофильного катализатора заключается в активации реагента и аналогична роли протона в кислотном катализе. Взаимодействие электрофильного катализатора с реагентом-основанием заключается в образовании донорно-акцепторной связи за счет пары электронов реагента-основания, занимающей вакантную орбиталь одного из атомов катализатора. Электрофильные катализаторы оказываются гораздо более эффективными, чем протонные кислоты при активировании таких слабоосновных реагентов, как галогены, алкилгалогениды, ангидриды и галогенангидриды, эпоксиды и т.п. Образование донорно-акцепторного комплекса либо сильно поляризует связь в молекуле реагента, либо приводит к полному ее разрыву с образованием катиона:


Образовавшийся катион или поляризованная молекула легко вступает в дальнейшее химическое превращение:


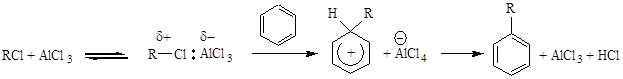
В некоторых случаях кислота Льюиса выполняет каталитические функции, активируя не сам реагент, а протонную кислоту, являющуюся сокатализатором:
Такие комплексы оказываются значительно более эффективными донорами протона (увеличение кислотности), чем исходные кислоты и относятся к разряду суперкислот (см. раздел Суреркислоты). В частности, комплекс HF×TaF5 обладает настолько высокой кислотностью, что отщепляет гидрид-анион от углеводородов с образованием карбокатиона:
Галогениды металлов (AlCl3, FeCl3, TiCl4, SnCl4, BF3) разлагаются при взаимодействии с кислород- или азотсодержащими органическими соединениями и водой либо образуют с ними очень прочные и малореакционноспособные комплексы. Поэтому галогениды металлов обычно не катализируют реакции с их участием.
ОСНОВНЫЙ КАТАЛИЗ
Основный катализ осуществляется веществами со свободной или лабильной электронной парой электронов, которые могут быть как анионами, так и нейтральными молекулами (HO — , RO — , NH2 — , R3N: и др.). При этом реагент должен обладать кислотными свойствами (по отношению к катализатору), и активирование реагента осуществляется за счет отрыва от него протона или образования водородной связи:
 или
или 
Причем в активации реагента могут принимать участия все формы оснований, образующиеся в реакционной массе, в том числе и ионы лиата, образующиеся по протолитической реакции катализатора с растворителем:
SH + B — ßà S — + BH
После активации реагента (уравнение (2.69)) превращения могут протекать по двум вариантам:
1) Мономолекулярная реакция. Из продуктов превращений на последующих стадиях регенерируется катализатор. Например:
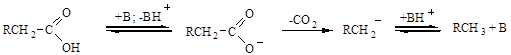
2)Бимолекулярная реакция. Участие активированного реагента в качестве нуклеофила в реакции со вторым реагентом. Эти реакции близки по механизму к нуклеофильному катализу (схема (2.27)). Отличие состоит в том, что при основном катализе протекает еще одна стадия в каталитическом цикле — отщепление катализатором протона от реагента с образованием сопряженного основания реагента, играющего далее роль нуклеофильного катализатора, но с обязательной регенерацией основного катализатора в конце каталитического цикла. Например в реакции альдольной конденсации:


В других случаях не происходит полного отщепления протона, а нуклеофильность реагента повышается за счет образования водородной связи с основанием-катализатором:

Механизмы и кинетика кислотно-основного катализа
Кислотно – основной катализ
Вы будете перенаправлены на Автор24
Общие положения кислотно — основного катализа
Кислотность и основность соединений определяет их реакционную способность.
Согласно определениям Бренстеда и Лоури, кислота — это вещество, отдающее протон, а основание — вещество, которое протон присоединяет. Донорами и акцепторами протонов могут выступать как электронейтральные молекулы, так и ионы. Сопряженные пары кислот и оснований изображает реакция:
где $AH$ — кислота;
Свойство кислот отдавать протон характеризуется константой кислотности $pK_a$. в водных растворах ее значение для протонных кислот лежит в широких пределах.
Примеры кислот и сопряженных с ними оснований: $N
По определению Льюиса кислотой является любое соединение, способное присоединить электронную пару, а основанием — соединение, способное электронную пару отдать. Тогда все электронодефицитные соединения будут являться кислотами $(AlCl_3$, $BF_3$, $S_nCl_4)$ или основаниями (фосфины, амины, сульфиды, карбанионы).
Кислотно — основный катализ вызван протолитической реакцией между кислотой и основанием и катализатором. Основная его особенность — переход протонов от катализатора к реагенту и обратно.
Так, при кислотном катализе протон переходит от катализатора к молекуле реагирующего вещества. При основном катализе первоначально катализатор служит акцептором протона или донором аниона по отношению к молекуле реагента.
С ростом константы диссоциации кислот и оснований активность катализаторов в кислотно — основном взаимодействии возрастает.
По типу кислотно — основного катализа протекают реакции
- гидратации;
- дегидратации;
- этерификации;
- гидролиза;
- поликонденсации и др.
Электрофильные и нуклеофильные реагенты
Электрофильность и нуклеофильность являются характеристиками реакционной способности веществ, которые участвуют в бимолекулярных гетеролитических реакциях. Одна из частиц, электрофил, выступает акцептором пары электронов, а другая — нуклеофил, донором электронов.
Скорость гетеролитической реакции будет тем больше, чем более ярко выражены электрофильные и нуклеофильные свойства.
Готовые работы на аналогичную тему
Электрофильные свойства увеличиваются при отщеплении протона от реагента основанием. Электрофильные и нуклеофильные свойства способны изменяться при комплексообразовании реагента с кислотой Льюиса.
Роль кислот и оснований в реакциях кислотно — основного типа как катализаторов заключается в том, что они способны образовывать с реагентом промежуточное соединение в результате донорно — акцепторного взаимодействия. В молекуле промежуточного соединения появляются реакционные центры, имеющие повышенную или пониженную электронную плотность, что способствует облегчению взаимодействия центрам с электрофильными или нуклеофильными молекулами второго реагента.
Классификация каталитических реакций кислотно — основного типа
Все реакции кислотно — основного типа в зависимости от природы частиц, выполняющих функции катализатора делят на группы:
Специфический кислотный катализ. Содержит реакции, катализируемые ионами водорода.
Гидролиз сложных эфиров в присутствии минеральных кислот. Включает присоединение протона к карбоксильному кислороду в результате донорно — акцепторного взаимодействия. Образовавшийся карбоний — катион обладает высокой электрофильностью, поэтому способен реагировать с водой. В ходе нескольких последовательных стадий образуется спирт, кислота и протон, способный вновь вступать в реакции.
Специфический основный катализ. Включает реакции, катализируемые ионами гидроксила. Промежуточными соединениями выступают анионы, образующиеся в результате присоединения основания или в результате отрыва катализатором протона от реагента.
Альдольная конденсация в присутствии гидроксил — ионов протекает в несколько этапов:
- отщепление протона и образование енолят — иона.
- присоединение енолят — иона к карбонильной группе другой молекулы.
- отщепление протона от растворителя, образование продукта реакции и регенерация катализатора.
Общий кислотно — основный катализ. Включает реакции, катализируемые кислотами и основаниями Бренстеда. В активации молекулы реагента принимают участие электрофильные частицы и кислоты Льюиса, которые выступают в качестве протона.
Реакция Фриделя — Крафтса (алкилирование ароматического кольца) катализируется хлоридом алюминия $AlCl_3$ в среде неполярного растворителя. Сначала образуется поляризованная молекула промежуточного соединения, возникает электрофильный реакционный центр. Этот центр вступает во взаимодействие с молекулой бензола с образованием $\sigma $- комплекса, один из атомов углерода переходит из состояния $sp^2$-гибридизации в $sp^3$-гибридизацию. Образуется продукт и катализатор.
Электрофильно — нуклеофильный катализ. Содержит реакции катализируемые донорами или акцепторами электронных пар.
Гидролиз сложного эфира в присутствии имидазола — типичный пример нуклеофильного катализа. Имидзол взаимодействует с эфиром и этим облегчает реакцию с водой. Молекулы промежуточного соединения сильно поляризуются и обладают большой реакционной способностью. В качестве нуклеофильных катализаторов могут выступать амины, ионы галогенов, некоторые анионы.
Зависимость константы скорости от функций кислотности
Реакции в органической химии могут катализироваться как кислотами, так и основаниями.
Экспериментально кислотно — основный катализ легко распознать по кривым зависимости логарифма константы скорости от $pH$, $H_0$ и других функций кислотности. Частный случай такой зависимости: двухстадийная реакция. Одна стадия ингибируется при добавлении кислоты, а другая в это время ускоряется.
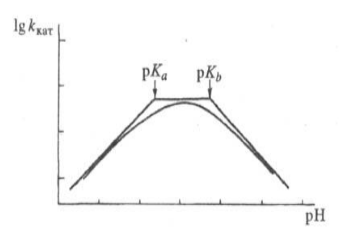
В реакции кетонов с гидроксиламином образование аминов будет замедляться в кислой среде. Это происходит из-за дезактивации гидроксиламина протонированием. Дегидратация, вторая стадия, катализируется кислотой и будет замедляться при высоких $pH$.
Получи деньги за свои студенческие работы
Курсовые, рефераты или другие работы
Автор этой статьи Дата последнего обновления статьи: 17 05 2021
http://lektsia.com/15x6ae9.html
http://spravochnick.ru/himiya/kisloty_i_osnovaniya/kislotno_osnovnoy_kataliz/