Основные стадии и кинетические особенности гетерогенных каталитических процессов
Гетерогенно-каталитическая реакция на поверхности твердого катализатора – это сложный многостадийный процесс. Наблюдаемая общая скорость каталитической реакции зависит от относительных скоростей нескольких различных по своей физической и химической природе стадий (ωΣ=Σωi).
Большинство известных промышленных каталитических реакций – это реакции между газообразными реагентами на твердых катализаторах.
1-я стадия. Внешняя диффузия. Как и в гетерогенном некаталитическом процессе, сначала происходит диффузия газообразного реагента из основного потока к внешней поверхности зерна катализатора через газовую пленку, в которой концентрация реагента ниже, а концентрация продукта выше, чем в основном потоке.
2-я стадия. Внутренняя диффузия. Основная часть молекул газообразного реагента диффундирует внутри пор катализатора. Скорость диффузии молекул через пористую среду во много раз меньше скорости их поступательного движения. Это объясняется тем, что во время прохождения через катализатор молекулы сталкиваются со стенками пор и с другими молекулами, что приводит к совершенно беспорядочному их движению.
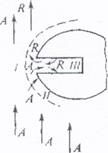 |
Схема участка зерна катализатора:
I – поток газа, обтекающий зерно катализатора;
II – пограничная газовая пленка;
Ш — — пора внутри катализатора;
A – исходный реагент; R – продукт реакции
3-я стадия – Активизированная адсорбция (Хемосорбции). Молекулы реагента адсорбируются на поверхности катализатора. Адсорбция представляет собой явление, связанное с уменьшением количества газа при соприкосновении газа (адсорбата) с твердым телом (адсорбентом), и заключается в некотором уплотнении газа на поверхности твердого тела. Различают физическую адсорбцию и хемосорбцию в зависимости от природы сил, вызывающих это концентрирование молекул адсорбата у поверхности твердого тела. Если эти силы имеют такую же природу, как и молекулярное взаимодействие в газах, жидкостях и твердых телах, говорят о физической адсорбции. При хемосорбции проявляются силы взаимодействия химической природы – молекулы адсорбата теряют свою индивидуальность, образуя поверхностные соединения с адсорбентом.
При протекании каталитических процессов основная роль принадлежит хемосорбцин, или активированной адсорбции, результатом которой является образование активированного комплекса адсорбции – неустойчивого промежуточного соединения между реагентом и катализатором. Стадия активированной адсорбции определяет специфичность действия катализаторов в отношении различных реакций. Если химическая связь реагента с адсорбентом слишком сильная, разрушение образовавшегося комплекса, ведущее к образованию продуктов, затрудняется. Если же связь адсорбента и адсорбата слишком слабая, близкая по своей природе к физической адсорбции, в молекуле адсорбата не происходит разрыхления связей, приводящих к снижению энергии активации каталитического процесса по сравнению с некаталитическим. При этом желательно, чтобы твердые катализаторы имели большую легко доступную поверхность, что достигается уменьшением размера зерен и увеличением их пористости. В ряде случаев внутренняя поверхность таких катализаторов достигает де-сятков и даже сотен квадратных метров на 1 см3 катализатора.
При наличии пористого катализатора реакция протекает как на внешней, так и на внутренней поверхности гранул катализатора. Часто внутренняя поверхность в тысячи раз превышает внешнюю поверхность, в этом случае влияние последней на процесс невелико.
4-я стадия – Поверхностная ХР. Вслед за адсорбцией происходит собственно поверхностная химическая реакция, которая заключается либо в перегруппировке активированного комплекса адсорбции, либо во взаимодействии одного адсорбированного реагента с молекулами другого. Механизм этой реакции может быть различным; от него зависит и вид кинетического уравнения. В результате поверхностной реакции образуется адсорбированный продукт.
5-я стадия – Десорбция продукта с поверхности катализатора. На этом этапе также проявляются специфические свойства катализатора: энергия связи адсорбированного продукта и адсорбента должна быть такой, чтобы десорбция в объем не вызывала затруднений.
Стадии 3, 4, 5 являются центральными в ходе каталитического процесса. Суммарно их можно рассматривать как поверхностную химическую реакцию. Эти стадии могут протекать одновременно с предыдущими – диффузионными – стадиями, причем как на внешней поверхности зерна катализатора, так и, в основном, на внутренней поверхности пор.
6-я стадия – Обратная внутренняя диффузия. Диффузия десорбированных газообразных продуктов из пор к внешней поверхности катализатора.
7-я стадия – Диффузия газообразных продуктов от поверхности катализатора в газовый поток через пограничную пленку, окружающую зерно катализатора.
В настоящее время установлено, что не вся поверхность катализатора однородна, поэтому катализ осуществляется только на так называемых активных центрах.
Механизм стадий катализа
3, 4, 5 стадии – Химические – Зависят от природы реагирующих веществ и природы катализатора.
Например, поверхность катализатора может адсорбировать лишь 1 реагент из нескольких. Стадии интенсифицируются путем увеличения температуры.
1 и 7 стадии – Области внешней диффузии – Интенсифицируются путем перемешивания реагирующих фаз.
2 и 6 стадии – Области внутренней диффузии – Интенсифицируются путем снижения размеров зерна и увеличения размеров пор, таким образом, чтобы внутренняя поверхность катализатора не уменьшалась.
КИНЕТИКА ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
В общем случае суммарное уравнение скорости всего процесса гетерогенного катализа должно включать описание каждой из его стадий. Но точно так же, как и при протекании гетерогенного некаталитического процесса, не все его стадии оказывают равное влияние на скорость катализа.
В большинстве случаев одна из стадий является наиболее медленной, лимитирующей процесс; она и определяет его скорость, поэтому для изыскания путей интенсификации такого процесса важно, прежде всего, установить лимитирующую стадию.
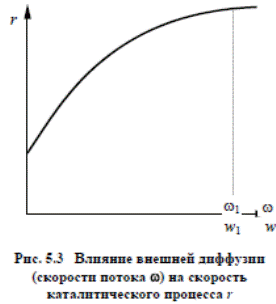
Из анализа гетерогенных некаталитических процессов известно, что если наиболее медленной стадией, лимитирующей общую скорость, является диффузионный перенос газообразного вещества через пограничный слой газа, т.е., если процесс протекает во внешнедиффузионной области, эффективным средством его ускорения служит увеличение скорости газового потока. На этом основан наиболее часто применяемый экспериментальный метод определения влияния диффузии на скорость каталитического процесса.
Для этой цели проводят серию опытов по определению скорости каталитической реакции при различной скорости потока реакционной смеси, но при постоянном отношении объема катализатора к объему смеси
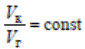 или
или
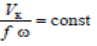
где Vк – объем катализатора; Vг – объем реакционной газовой смеси; f – площадь сечения контактной трубки; ω – линейная скорость газового потока.
При увеличении ω (что достигается уменьшением f) скорость реакции
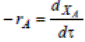
будет возрастать только до тех пор, пока процесс протекает во внешнедиффузионной области (рис. 5.3).
На участке кривой от ω = 0 до ω = ω1 скорость потока оказывает влияние на скорость реакции и, следовательно, процесс протекает во внешнедиффузионной области.
Влияние внутренней диффузии исследуют путем проведения серии опытов при скорости потока ω >ω1 (в области, где внешняя диффузия уже не оказывает влияния на общую скорость процесса). Опыты проводят на зернах катализатора различного размера, результаты опытов выражают в виде графической зависимости 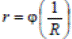 . На графике эта зависимость имеет такой же характер, что и для некаталитических процессов в системе г – т (рис. 5.4).
. На графике эта зависимость имеет такой же характер, что и для некаталитических процессов в системе г – т (рис. 5.4).
Активное действие катализатора обусловлено прежде всего предварительной адсорбцией реагирующих веществ на поверхности катализатора (стадии III – V на рис.5.2), что оказывает большое влияние на скорость гетерогенного катализа.
Адсорбция является самопроизвольным процессом, поэтому
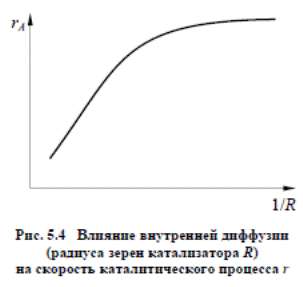
она сопровождается убылью энергии системы и связана с выделением тепла. Существует два вида адсорбции – физическая и химическая (последняя называется также активированной адсорбцией, или хемосорбцией). Процесс катализа связан с хемосорбцией, но хемосорбция может быть обратимой и необратимой. Естественно, что в процессе катализа хемосорбция должна быть обратимой, так как активные центры должны непрерывно возобновлять свою функциональную деятельность по отношению к реагентам. Необратимая адсорбция вызывает отравление катализатора.
С целью установления функциональной зависимости скорости гетерогенного катализа от различных факторов проводят всесторонние исследования, что позволяет определить влияние различных показателей на скорость адсорбции и десорбции реагентов поверхностью катализатора, а также на скорость протекания других стадий, определяющих каталитический процесс в целом. Полученные при этом данные используют для составления теоретических уравнений, позволяющих установить общие закономерности в идеальных условиях.
Нахождение кинетических уравнений и определение оптимальных параметров является главной целью научных исследований в области каталитических процессов, так как эти данные используются затем для расчета каталитических реакторов.
Температура оказывает весьма существенное влияние на каталитические процессы, так как при повышении температуры увеличивается константа скорости реакции и одновременно изменяется константа равновесия. Для процессов, проходящих в кинетической области, повышение температуры всегда способствует приближению процесса к состоянию равновесия. Но, как известно, при обратимых реакциях равновесная степень превращения X* при повышении Т уменьшается для экзотермических реакций и увеличивается для эндотермических реакций. Поэтому закономерности, отражающие суммарную скорость реакции и действительную степень превращения для экзотермических и эндотермических реакций, совершенно различны. При этом наблюдается такая же функциональная зависимость Х = f (Т), как и для некаталитических процессов.
Время контакта (время соприкосновения) реагирующих веществ с катализатором также является важной технологической характеристикой каталитического процесса, так как оно определяет его интенсивность. При расчете реакторов время контакта определяют по уравнению 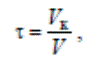 где τ – время контакта; Vк – объем катализатора; V – объем реакционной смеси, проходящей через катализатор в единицу времени.
где τ – время контакта; Vк – объем катализатора; V – объем реакционной смеси, проходящей через катализатор в единицу времени.
Величина, обратная времени контакта, называется объемной скоростью и выражается уравнением
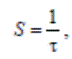 где S – объемная скорость (объем реакционной смеси, проходящей через единицу объема катализатора в единицу времени), м 3 (газа) / м 3 (катализ) с 1 = с –1 .
где S – объемная скорость (объем реакционной смеси, проходящей через единицу объема катализатора в единицу времени), м 3 (газа) / м 3 (катализ) с 1 = с –1 .
При увеличении объемной скорости обычно снижается степень превращения, однако при этом возрастает интенсивность работы аппарата, т.е. увеличивается количество целевого продукта, получаемого с единицы объема катализатора в единицу времени. Это объясняется тем, что при увеличении скорости потока реагирующая система в большей мере удалена от равновесия, процесс протекает в области высоких скоростей за счет большой движущей силы.
Интенсивность катализатора выражают в виде уравнения
где G – производительность катализатора, кг / ч м 3 ; ρ – плотность реагента при нормальных условиях, кг / м 3 (для аммиака ρ = 0,7708 кг м–3); z – мольная доля целевого продукта в газовой смеси; S – объемная скорость, ч –1 .
Из уравнения видно, что при увеличении объемной скорости производительность катализатора возрастает. Однако возможности для увеличения S ограничены, так как при этом степень превращения ХА уменьшается (снижается концентрация целевого продукта, что затрудняет выделение его из реакционной смеси), возрастает расход энергии и нарушается автотермичность экзотермической реакции вследствие относительного увеличения объема реакционной смеси.
Если проводить экзотермическую реакцию с различным временем соприкосновения реакционной смеси с катализатором τ, получают серию кривых на осях координат Т – ХА с максимальным значением Хmax, что позволяет установить оптимальную температурную последовательность, обеспечивающую наиболее эффективное ведение процесса. Его следует начинать при высокой температуре, когда скорость процесса велика и, следовательно, достигается высокая интенсивность, а затем вести процесс при снижении температуры и увеличении времени соприкосновения реагирующих масс, при этом возрастает значение ХА
Применение давления является одним из способов повышения степени превращения при промышленном осуществлении обратимых каталитических реакций, проходящих с уменьшением объема. Давление становится решающим фактором, когда активность катализатора и величина X* низкие.
Химическая кинетика. Скорость химических реакций
Темы кодификатора ЕГЭ: Скорость реакции. Ее зависимость от разных факторов.
Скорость химической реакции показывает, как быстро происходит та или иная реакция. Взаимодействие происходит при столкновении частиц в пространстве. При этом реакция происходит не при каждом столкновении, а только когда частица обладают соответствующей энергией.
Скорость реакции – количество элементарных соударений взаимодействующих частиц, заканчивающихся химическим превращением, за единицу времени.
Определение скорости химической реакции связано с условиями ее проведения. Если реакция гомогенная – т.е. продукты и реагенты находятся в одной фазе – то скорость химической реакции определяется, как изменение концентрации вещества в единицу времени:
υ = ΔC / Δt
Если реагенты, или продукты находятся в разных фазах, и столкновение частиц происходит только на границе раздела фаз, то реакция называется гетерогенной, и скорость ее определяется изменением количества вещества в единицу времени на единицу реакционной поверхности:
υ = Δν / (S·Δt)
Факторы, влияющие на скорость химической реакции
1. Температура
Самый простой способ изменить скорость реакции – изменить температуру . Как вам, должно быть, известно из курса физики, температура – это мера средней кинетической энергии движения частиц вещества. Если мы повышаем температуру, то частицы любого вещества начинают двигаться быстрее, а следовательно, сталкиваться чаще.
Однако при повышении температуры скорость химических реакций увеличивается в основном благодаря тому, что увеличивается число эффективных соударений. При повышении температуры резко увеличивается число активных частиц, которые могут преодолеть энергетический барьер реакции. Если понижаем температуру – частицы начинают двигаться медленнее, число активных частиц уменьшается, и количество эффективных соударений в секунду уменьшается. Таким образом, при повышении температуры скорость химической реакции повышается, а при понижении температуры — уменьшается .
Обратите внимание! Это правило работает одинаково для всех химических реакций (в том числе для экзотермических и эндотермических). Скорость реакции не зависит от теплового эффекта. Скорость экзотермических реакций при повышении температуры возрастает, а при понижении температуры – уменьшается. Скорость эндотермических реакций также возрастает при повышении температуры, и уменьшается при понижении температуры.
Более того, еще в XIX веке голландский физик Вант-Гофф экспериментально установил, что скорость большинства реакций примерно одинаково изменяется (примерно в 2-4 раза) при изменении температуры на 10 о С.
Правило Вант-Гоффа звучит так: повышение температуры на 10 о С приводит к увеличению скорости химической реакции в 2-4 раза (эту величину называют температурный коэффициент скорости химической реакции γ).
Точное значение температурного коэффициента определяется для каждой реакции.

здесь v2 — скорость реакции при температуре T2,
v1 — скорость реакции при температуре T1,
γ — температурный коэффициент скорости реакции, коэффициент Вант-Гоффа.
В некоторых ситуациях повысить скорость реакции с помощью температуры не всегда удается, т.к. некоторые вещества разлагаются при повышении температуры, некоторые вещества или растворители испаряются при повышенной температуре, т.е. нарушаются условия проведения процесса.
2. Концентрация
Также изменить число эффективных соударений можно, изменив концентрацию реагирующих веществ . Понятие концентрации, как правило, используется для газов и жидкостей, т.к. в газах и жидкостях частицы быстро двигаются и активно перемешиваются. Чем больше концентрация реагирующих веществ (жидкостей, газов), тем больше число эффективных соударений, и тем выше скорость химической реакции.
На основании большого числа экспериментов в 1867 году в работах норвежских ученых П. Гульденберга и П. Вааге и, независимо от них, в 1865 году русским ученым Н.И. Бекетовым был выведен основной закон химической кинетики, устанавливающий зависимость скорости химической реакции от концентрации реагирующих веществ:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ в степенях, равных их коэффициентам в уравнении химической реакции.
Для химической реакции вида: aA + bB = cC + dD закон действующих масс записывается так:

здесь v — скорость химической реакции,
CA и CB — концентрации веществ А и В, соответственно, моль/л
k – коэффициент пропорциональности, константа скорости реакции.
Например , для реакции образования аммиака:
закон действующих масс выглядит так:

Константа скорости реакции k показывает, с какой скоростью будут реагировать вещества, если их концентрации равны 1 моль/л, или их произведение равно 1. Константа скорости химической реакции зависит от температуры и не зависит от концентрации реагирующих веществ.
В законе действующих масс не учитываются концентрации твердых веществ, т.к. они реагируют, как правило, на поверхности, и количество реагирующих частиц на единицу поверхности при этом не меняется.
В большинстве случаев химическая реакция состоит из нескольких простых этапов, в таком случае уравнение химической реакции показывает лишь суммарное или итоговое уравнение происходящих процессов. При этом скорость химической реакции сложным образом зависит (или не зависит) от концентрации реагирующих веществ, полупродуктов или катализатора, поэтому точная форма кинетического уравнения определяется экспериментально, или на основании анализа предполагаемого механизма реакции. Как правило, скорость сложной химической реакции определяется скоростью его самого медленного этапа (лимитирующей стадии).
3. Давление
Концентрация газов напрямую зависит от давления . При повышении давления повышается концентрация газов. Математическое выражение этой зависимости (для идеального газа) — уравнение Менделеева-Клапейрона:
pV = νRT
Таким образом, если среди реагентов есть газообразное вещество, то при повышении давления скорость химической реакции увеличивается, при понижении давления — уменьшается .
Например. Как изменится скорость реакции сплавления извести с оксидом кремния:
при повышении давления?
Правильным ответом будет – никак, т.к. среди реагентов нет газов, а карбонат кальция – твердая соль, нерастворимая в воде, оксид кремния – твердое вещество. Газом будет продукт – углекислый газ. Но продукты не влияют на скорость прямой реакции.
4. Катализатор
Еще один способ увеличить скорость химической реакции – направить ее по другому пути, заменив прямое взаимодействие, например, веществ А и В серией последовательных реакций с третьим веществом К, которые требуют гораздо меньших затрат энергии (имеют более низкий активационный энергетический барьер) и протекают при данных условиях быстрее, чем прямая реакция. Это третье вещество называют катализатором .

Катализаторы – это химические вещества, участвующие в химической реакции, изменяющие ее скорость и направление, но не расходующиеся в ходе реакции (по окончании реакции не изменяющиеся ни по количеству, ни по составу). Примерный механизм работы катализатора для реакции вида А + В можно представить так:
A + K = AK
AK + B = AB + K
Процесс изменения скорости реакции при взаимодействии с катализатором называют катализом. Катализаторы широко применяют в промышленности, когда необходимо увеличить скорость реакции, либо направить ее по определенному пути.
По фазовому состоянию катализатора различают гомогенный и гетерогенный катализ.
Гомогенный катализ – это когда реагирующие вещества и катализатор находятся в одной фазе (газ, раствор). Типичные гомогенные катализаторы – кислоты и основания. органические амины и др.
Гетерогенный катализ – это когда реагирующие вещества и катализатор находятся в разных фазах. Как правило, гетерогенные катализаторы – твердые вещества. Т.к. взаимодействие в таких катализаторах идет только на поверхности вещества, важным требованием для катализаторов является большая площадь поверхности. Гетерогенные катализаторы отличает высокая пористость, которая увеличивает площадь поверхности катализатора. Так, суммарная площадь поверхности некоторых катализаторов иногда достигает 500 квадратных метров на 1 г катализатора. Большая площадь и пористость обеспечивают эффективное взаимодействие с реагентами. К гетерогенным катализаторам относятся металлы, цеолиты — кристаллические минералы группы алюмосиликатов (соединений кремния и алюминия), и другие.

Пример гетерогенного катализа – синтез аммиака:
В качестве катализатора используется пористое железо с примесями Al2O3 и K2O.
Сам катализатор не расходуется в ходе химической реакции, но на поверхности катализатора накапливаются другие вещества, связывающие активные центры катализатора и блокирующие его работу (каталитические яды). Их необходимо регулярно удалять, путем регенерации катализатора.
В биохимических реакция очень эффективными оказываются катализаторы – ферменты. Ферментативные катализаторы действуют эффективно и избирательно, с избирательностью 100%. К сожалению, ферменты очень чувствительны к повышению температуры, кислотности среды и другим факторам, поэтому есть ряд ограничений для реализации в промышленных масштабах процессов с ферментативным катализом.
Катализаторы не стоит путать с инициаторами процесса и ингибиторами.
Например , для инициирования радикальной реакции хлорирования метана необходимо облучение ультрафиолетом. Это не катализатор. Некоторые радикальные реакции инициируются пероксидными радикалами. Это также не катализаторы.
Ингибиторы – это вещества, которые замедляют химическую реакцию. Ингибиторы могут расходоваться и участвовать в химической реакции. При этом ингибиторы не являются катализаторами наоборот. Обратный катализ в принципе невозможен – реакция в любом случае будет пытаться идти по наиболее быстрому пути.
5. Площадь соприкосновения реагирующих веществ
Для гетерогенных реакций одним из способов увеличить число эффективных соударений является увеличение площади реакционной поверхности . Чем больше площадь поверхности контакта реагирующих фаз, тем больше скорость гетерогенной химической реакции. Порошковый цинк гораздо быстрее растворяется в кислоте, чем гранулированный цинк такой же массы.
В промышленности для увеличения площади контактирующей поверхности реагирующих веществ используют метод «кипящего слоя».
Например , при производстве серной кислоты методом «кипящего слоя» производят обжиг колчедана.
6. Природа реагирующих веществ
На скорость химических реакций при прочих равных условиях также оказывают влияние химические свойства, т.е. природа реагирующих веществ.
Менее активные вещества будут имеют более высокий активационный барьер, и вступают в реакции медленнее, чем более активные вещества.
Более активные вещества имеют более низкую энергию активации, и значительно легче и чаще вступают в химические реакции.
Более стабильные вещества — это, например, те вещества, которые окружают нас в быту, либо существуют в природе.
Например , хлорид натрия NaCl (поваренная соль), или воды H2O, или металлическое железо Fe.
Более активные вещества мы можем встретить в быту и природе сравнительно редко.
Например , оксид натрия Na2O или сам натрий Na в быту и в природе не не встречаем, т.к. они активно реагируют с водой.
При небольших значениях энергии активации (менее 40 кДж/моль) реакция проходит очень быстро и легко. Значительная часть столкновений между частицами заканчивается химическим превращением. Например, реакции ионного обмена происходят при обычных условиях очень быстро.
При высоких значениях энергии активации (более 120 кДж/моль) лишь незначительное число столкновений заканчивается химическим превращением. Скорость таких реакций пренебрежимо мала. Например, азот с кислородом практически не взаимодействует при нормальных условиях.
При средних значениях энергии активации (от 40 до 120 кДж/моль) скорость реакции будет средней. Такие реакции также идут при обычных условиях, но не очень быстро, так, что их можно наблюдать невооруженным глазом. К таким реакциям относятся взаимодействие натрия с водой, взаимодействие железа с соляной кислотой и др.
Вещества, стабильные при нормальных условиях, как правило, имеют высокие значения энергии активации.
Кинетическое уравнение скорости гетерогенной каталитической реакции
Тогда уравнение скорости между протонированной формой A и Y выразится как

Уравнение (13) отражает противоборствующее влияние кислотности среды на скорость реакции: 1-й член увеличивается с ростом кислотности, но 2-й уменьшается. Такой характер влияния обусловливает экстремальную зависимость скорости соответствующих реакций от кислотности среды. Подобные зависимости наблюдаются для кислотно-каталитических реакций карбонильных соединений с азотистыми основаниями.
Гетерогенный катализ важнейшая область катализа, имеющая широкое практическое приложение. Достаточно в этом плане отметить такие важные процессы как синтез аммиака



В то же время гетерогенно-каталитические реакции – сложные и многомерные физико-химические системы, представляющие большой научный интерес.
Преимущества гетерогенного катализа перед гомогенным состоят в малом расходе катализатора на единицу количества продукта, снижении или полном устранении токсичных сточных вод и расходов дополнительных реагентов на промывку реакционной массы. Это обусловлено тем, что катализатор и реакционная масса находятся в разных фазах.
По способу осуществления гетерогенно-каталитические процессы делятся на процессы со стационарным (неподвижным) катализатором, где он используется в виде достаточно крупных гранул (0,3 – 1 см) и на процессы с подвижным катализатором (плавающим, диспергированным или псевдоожиженным). В последнем случае катализатор применяют только в диспергированном виде, когда он способен перемешиваться под влиянием потока реакционной массы.
Каталитические процессы осуществляются в газовой или жидкой фазе.
В зависимости от типа механизма реакций, протекающих на гетерогенных катализаторах последние подразделяют на следующие группы:
1. Ионные, под влиянием которых протекают реакции по ионному механизму.
2. Электронные, катализирующие гомолитические реакции
3. Бифункциональные, совмещающие ионный и электронный катализ
К ионным катализаторам относятся следующие:
1. Кислотно-основные катализаторы
В группу этих катализаторов входят
а) оксиды некоторых металлов, например Al 2 O 3 , W 2 O 3 , ThO 2 .
б) нейтральные и кислые соли: Ca 3 ( PO 4 )2, CaHPO 4 , MgHPO 4 , а также природные и синтетические алюмосиликаты ( Al 2 O 3 ) m ( SiO 2 ) n ( H 2 O ) p .
в) Протонные и апротонные кислоты на носителях ( H 3 PO 4 на Al 2 O 3 или кизельгуре, BF 3 на Al 2 O 3 , гетерополикислоты) и ионообменные смолы.
2. Комплексообразующие соли переходных металлов на носителях: Cu 2 Cl 2 , HgCl 2 , PdCl 2 + CuCl 2 , CuC º C С u , NiCl 2 и другие.
Все катализаторы ионного типа являются изоляторами или ионными проводниками электрического тока. Наиболее распространены катализаторы кислотного типа, являющиеся протонными (бренстедовскими) или апротонными (льюисовскими) кислотами.
Ионообменные смолы используют главным образом для катализа жидкофазных реакций, которые и в гомогенных условиях катализируются протонными кислотами (дегидратация, этерификация, алкилирование, конденсация по карбонильной группе) и только до температуры » 150 ° C , выше которой ионообменные смолы склонны к деструкции. Для тех же но газофазных реакций и гидратации олефинов используют фосфорную кислоту на носителях. Оксиды и соли применяются как для перечисленных выше реакций, так и для крекинга углеводородов (алюмосиликат) или алкилирования аминов ( Al 2 O 3 ) Соли обладают мягким действием (например, фосфаты), что обусловило их использование для тех реакций, реагенты или продукты которых чувствительны к влиянию более активных катализаторов.
Гетерогенные катализаторы гомолитических реакций всегда являются электронными проводниками тока или полупроводниками, они включают следующие группы веществ:
1. Перехолные металлы I Б и VIII групп ( Cu , Ag , Fe , Ni , Co , Pt , Pd ) периодической системы Д. И. Менделеева.
2. Оксиды металлов ( MgO , ZnO , CuO , Fe 2 O 3 , Cr 2 O 3 , WO 3 , MoO 3 , V 2 O 5 ) сульфиды ( WS 3 , MoS 3 ) и смеси, содержащие один основной оксид с небольшими добавками других (модифицированные оксидные катализаторы)
3. Сложные оксидные и сульфидные катализаторы с соизмеримым соотношением компонентов, а также соли-полупроводники: хромиты ( CrO × Cr 2 O 3 , ZnO × Cr 2 O 3 ), вольфраматы ( CoO × WO 3 ), молибдаты ( Bi 2 O 3 × MoO 3 , NiS × MoS 3 ) ванадаты и другие.
Между ионными и электронными катализаторами не существует резкой границы, некоторые из них способны ускорять ионные и гомолитические реакции. Из индивидуальных веществ это особенно относится к оксидам, часть из которых катализирует ионные, а остальные – гомолитические реакции. Относительная активность оксидных катализаторов в ионных и гомолитических реакциях может быть проиллюстрирована на примере конкуренции двух реакций – ионной реакции дегидратации этанола и гомолитический реакции – его дегидрирования.
относительная доля продукта дегидратации, %
относительная доля продукта дегидратации, %
Из приведенных данных видно, что некоторые оксиды являются бифункциональными катализаторами. Бифунциональности катализаторов можно также достичь используя смеси оксидов разного типа. Лучшим примером является система ZnO на Al 2 O 3 , успешно применявшаяся для синтеза бутадиена из этанола, который представляет собой совмещенный процесс дегидратации, димеризации и дегидрирования. Другой пример бифункциональных катализаторов – металлы платиновой группы на носителях кислотного типа ( Al 2 O 3, алюмосиликаты), являются катализаторами каталитического риформина в котором протекают гомолитические реакции дегидрирования и ионные реакции изомеризации и расщепления.
Гетерогенные катализаторы должны удовлетворять следующим требованиям:
— высокая каталитическая активность
— достаточно большая селективность (избирательность) в отношении целевой реакции
— простота получения, обеспечивающая воспроизводимость всех свойств катализатора.
— высокая механическая прочность к сжатию, удару и истиранию
— достаточная стабильность всех свойств катализатора на протяжении его службы и способность к их восстановлению при том или ином методе регенерации.
— небольшие экономические затраты на катализатор при производстве единицы продукции.
Обеспечение этих требований достигается главным образом при разработке состава катализатора и способа его получения.
Гетерогенные катализаторы сравнительно редко применяются в индивидуальном виде и часто содержат различные добавки, получившие название модификаторов. Цели их введения разнообразны: повышение активности катализатора (промоторы), избирательности и стабильности работы, улучшение механических или структурных свойств. Фазовые и структурные модификации стабилизируют соответственно активную фазу твердого катализатора или пористую структуру его поверхности. Так, в меди-хромистых катализаторах гидрирования оксид хрома препятствует восстановдению оксида меди с превращением его в неактивную форму. Добавление уже 1% Al 2 O 3 к железному катализатору значительно увеличивает его поверхность, препятствуя спеканию и закрытию пор. Некоторые модификаторы существенно повышают стабильность работы катализатора.
В смешанных катализаторах, где компоненты находятся в соизмеримых количествах, могут образовываться новые более активные соединения, их твердые растворы в основном компоненте или многофазные системы, обладающие специфическим каталитическим действием. При этом свойства смешанного катализатора не являются простой суммой свойств его компонентов. Это приводит к заметному увеличению избирательности, активности и других свойств катализатора.
К числу модификаторов можно отнести и носители (трегеры), особенно часто применяемые в случае дорогостоящих металлических катализаторов ( Pt , Pd , Ni , Co ). Роль последнего состоит в повышении активной поверхности, увеличении термостойкости и механической прочности катализатора и т.д. В качестве последнего используют алюмосиликаты, Al 2 O 3 , Cr 2 O 3 , SiO 2 , активированный уголь, пемзу, кизельгур и другие природные и синтетические материалы.
Как правило, гетерогенно-каталитический процесс протекает через ряд последовательных, а иногда и последовательно параллельных стадий, существенно различающихся по своей природе. К таким процессам относятся:
1. диффузия реагентов из потока к внешней поверхности зерна катализатора.
2. диффузия реагентов к внутренней поверхности зерна катализатора (в поры)
3. адсорбция реагентов на поверхности катализатора
4. собственно химическая реакция
5. десорбция продуктов реакции с поверхности катализатора
6. диффузия продуктов с внутренней поверхности зерна катализатора
7. диффузия продуктов с внешней поверхности зерна катализатора в поток
Любая из этих стадий может оказаться лимитирующей и, следовательно, определять скорость процесса в целом. Таким образом, кинетические закономерности гетерогенно-каталитического процесса могут контролироваться закономерностями диффузии, адсорбции или химической реакции, а в пограничных случаях – их совокупностью. Это определяет специфику кинетики гетерогенного катализа по сравнению с кинетикой гомогенного катализа.
Принято различать кинетически- и диффузионно- контролируемые области протекания гетерогенно-каталитических процессов. В первых из них общая скорость процесса определяется скоростью химической реакции на поверхности, во вторых – диффузией реагентов или продуктов реакции. Кроме того, существуют области, контролируемые сорбцией реагентов или десорбцией продуктов.
Более детально различают следующие 5 основных областей:
1. Внешнедиффузионная область. В этой области скорость в целом определяется скоростью транспорта реагентов из потока к внешней поверхности зерна катализатора (или скоростью диффузии продуктов с внешней поверхности в поток)
2. Внутридиффузионная область. В этом случае скорость процесса лимитируется диффузией реагентов от внешней поверхности зерна катализатора к его внутренней поверхности (или наоборот, диффузией продуктов реакции из пор катализатора к его внешней поверхности)
3. Внешнекинетическая область. В этом случае скорость процесса лимитируется скоростью химической реакции на внешней поверхности зерна катализатора. Это возможно, если скорость химической реакции на внешней поверхности значительно превосходит скорость внутренней диффузии (стадии 2 и 6), но значительно меньше скорости внешней диффузии.
4. Внутрикинетическая область. Скорость процесса определяется скоростью химической реакции, причем последняя протекает на внешней и внутренней поверхности зерна катализатора. Это возможно, когда химическая реакция идет значительно медленнее внешней и внутренней диффузии.
5. Сорбционная область. Здесь скорость процесса определяется адсорбцией реагентов или десорбцией продуктов.
Строгие границы между этими областями отсутствуют, они перекрываются переходными областями, в которых сочетаются закономерности различных областей.
При экспериментальном исследовании и расчетах гетерогенно-каталитических процессов важно знать область, в которой протекает реакция, отчего зависит вид описывающих её кинетических уравнений. Так, при лимитировании скорости процесса химической реакцией на всей поверхности катализатора (внутрикинетическая область) скорость диффузии не играет роли, и результаты процесса не будут зависеть от размера зерен катализатора. Наоборот: во внешне- или внутридиффузионной области размер зерна играет большую роль, так как скорость диффузии на единицу массы катализатора зависит от величины внешней поверхности, которая определяет и диффузию в поры. Таким образом, проводя серию экспериментов с катализатором разного размера зерна и наблюдая за изменением конверсии, можно различить кинетическую и диффузионную области, а также определить размер зерна катализатора, необходимый для достижения кинетической области.
Тестом на различие внешне- и внутридиффузионной областей является зависимость скорости внешней диффузии от линейной скорости газового потока, тогда как внутренняя диффузия не зависит от этого параметра. Линейную скорость потока можно варьировать, меняя объем катализатора, соблюдая при этом постоянство времени контакта.

Легко убедиться, что такое требование можно соблюсти при условии пропорционального изменения объема υ линейной скорости n , связанной с объемной скоростью потока соотношением

Изменение или постоянство степени конверсии в таких опытах свидетельствует о наличии или отсутствии внешнедиффузионного торможения.
При исследовании гетерогенно-каталитических реакций наиболее достоверные данные получаются в проточно-циркуляционных установках, хотя часто используют метод исследования в потоке.
Во избежание нарушений в режиме потока реагентов диаметр каталитической ячейки или трубы должен быть равным не менее 6-7 диаметров зерна катализатора.
Обычно при работе катализаторов происходит постепенное изменение их активности и селективности. Поэтому кинетический эксперимент можно ставить лишь в период их стационарных свойств, который наступает после более или менее длительного холостого пробега и заканчивается при наличии признаков заметной дезактивации катализаторов.
Кинетические эксперименты проводят при варьировании начальных концентраций (парциальных давлений) реагентов и продуктов, а также условного времени контакта.
При кинетическом анализе реакций на поверхности катализатора руководствуются законом действующих поверхностей, согласно которому скорость химической реакции пропорциональна поверхностной концентрации реагентов, участвующих в акте химического взаимодействия.
В случае мономолекулярных реакций, например A ® B , акт химического взаимодействия можем представлять собой превращение вещества, сорбированного на активном центре поверхности, или его взаимодействия с соседним свободным центром поверхности. Эти двум механизмам соответствуют кинетические уравнения

В случае бимолекулярных реакций, например A + Y ® B , возможны также два механизма. Согласно одному из них два реагента адсорбируются на разных активных центрах поверхности и взаимодействуют между собой, если эти центры находятся в достаточной близости друг от друга (механизм Хиншельвуда). Такому механизму соответствует следующее кинетическое уравнение

Согласно другому механизму на поверхности сорбируется один из реагентов, активирования которого достаточно для взаимодействия с налетающей из объема молекулой второго реагента (“ударный” механизм Ридила). Такому механизму соответствует кинетическое уравнение

Неэлементарные реакции состоят из ряда элементарных стадий, составляющих их механизм. Кинетика таких реакций определяется последовательностью элементарных стадий, их характером (обратимые, необратимые), природой реагентов, интермедиатов и продуктов реакции. При кинетическом анализе неэлементарных реакций возникает задача определения концентраций интермедиатов, играющих ключевую роль в образовании продуктов или расходовании реагентов. В качестве инструмента такого определения используется принцип квазистационарных концентраций Боденштейна – Семенова. Согласно этому принципу скорость изменения концентраций нестабильных интермедиатов пренебрежимо мала по сравнению со скоростью изменения концентраций реагентов и продуктов реакции и её можно считать равной нулю. Применение принципа стационарных концентраций к неэлементарным реакциям, протекающим по сложному механизму, позволяет исключить из кинетического описания процессов неизвестные концентрации интермедиатов и получить одно или некоторый минимум дифференциальных уравнений скорости, выраженных через подлежащие измерению концентрации реагентов и продуктов реакции.
Рассмотрим пример неэлементарной реакции, описываемой стехиометрией

и протекающей через образование интермедиата Q


Скорость реакции можно приравнять к скорости образования продукта B

В соответствии с принципом квазистационарных концентраций

Откуда 
Подставляя последнее выражение в уравнение (1) приходим к уравнению скорости реакции

Если экспериментально возможно непосредственно измерить скорость реакции, то обработку кинетических данных можно провести, преобразуя уравнение (3) как:

Последнее уравнение приводится к виду

Обрабатывая зависимость (4) в координатах  по ординате находят k 1 , а по тангенсу угла наклона
по ординате находят k 1 , а по тангенсу угла наклона  . Полученных констант достаточно для кинетического описания реакции, так как, разделив числитель и знаменатель уравнения (3) на k 2 , приходят к уравнению
. Полученных констант достаточно для кинетического описания реакции, так как, разделив числитель и знаменатель уравнения (3) на k 2 , приходят к уравнению

с известными коэффициентами.
Плодотворность метода квазистационарных концентраций может быть проиллюстрирована на примере химических реакций, катализируемых ферментами.

Где Е – активный центр фермента, Х – промежуточное соединение, которое часто называют фермент-субстратным компонентом.
Кинетическая модель этой реакции может быть представлена системой кинетических уравнений


Решение этой системы уравнений в аналитической форме невозможно, поэтому оценка констант уравнений k 1 , k 2 , k 3 и k 4 производится численными методами.
Ферментативные реакции обычно изучают при концентрациях фермента значительно меньших, чем концентрациях субстрата, поэтому можно с высокой степенью приближения принять, что ферментативная реакция протекает стационарно, так что  . Поскольку [ E ]0 = [ E ] + [ X ] где [ E ]0 – начальная концентрация фермента (точнее, его активных центров), уравнение (6) можно представить в виде
. Поскольку [ E ]0 = [ E ] + [ X ] где [ E ]0 – начальная концентрация фермента (точнее, его активных центров), уравнение (6) можно представить в виде
 Разрешая последнее уравнение относительно [ X ], имеем
Разрешая последнее уравнение относительно [ X ], имеем

Вводя обозначение  ,
, 
Приводим уравнение (8) к форме

Подставляя полученное выражение в уравнение (7) с учетом равенства [ E ]0 = [ E ] + [ X ] приходим к уравнению

Уравнение (9) называется уравнением Михаэлиса – Ментен. При низких конверсиях концентраций продукта P в уравнении (9) можно пренебречь и последнее приобретает вид

При [ S ] KS можно пренебречь единицей в знаменателе уравнения (10) и скорость реакции имеет первый порядок по субстрату:

В противоположном случае [ S ] KS можно пренебречь вторым членом в знаменателе уравнения (10) и реакция имитирует нулевой порядок по субстрату

В обоих случаях порядок реакции является первым по концентрации активных центров фермента. Величина u S = k 3 [ E ]0 называется максимальной скоростью ферментативной реакции, а k 3 – числом реакционных центров для прямой реакции. Величина  называется константой Михаэлиса для субстратов. Как видно из уравнения (10) она равна концентрации субстрата, при которой скорость реакции составляет половину максимальной. Значение u S и KS можно найти, преобразуя уравнение (10) в линеаризированную форму
называется константой Михаэлиса для субстратов. Как видно из уравнения (10) она равна концентрации субстрата, при которой скорость реакции составляет половину максимальной. Значение u S и KS можно найти, преобразуя уравнение (10) в линеаризированную форму

Тогда u S определяется по ординате линейной зависимости  от
от  , а KS – по тангенсу угла наклона с учетом найденной величины u S .
, а KS – по тангенсу угла наклона с учетом найденной величины u S .
Параметры u P и KP – могут быть найдены численными методами с учетом известных значений u S и KS путем обработки экспериментальных данных полученных при достаточно высоких конверсиях субстрата.
http://chemege.ru/kinetika/
http://www.trotted.narod.ru/physchem/27.htm