Зависимость фермента от количества субстрата описывает ферментативная кинетика
Уравнения Михаэлиса-Ментен и Лайнуивера-Берка
Общую теорию ферментативной кинетики и зависимость активности фермента от субстрата.описали Л.Михаэлис и М.Л.Ментен, выразив его в своем уравнении. Бриггс и Холдейн усовершенствовали их уравнение, введя введя в него константу Михаэлиса (Km), определяемую экспериментально.
Уравнение Михаэлиса-Ментен показывает взаимосвязь максимально возможной скорости, реальной скорости реакции, константы Михаэлиса и концентрации субстрата. Так как пользоваться графиком, построенным в прямых координатах V и [S] для точных расчетов неудобно, то Г.Лайнуивер и Д.Бэрк преобразовали уравнение Бриггса–Холдейна в обратные координаты.
Уравнение Михаэлиса-Ментен
Уравнение Лайнуивера-Бэрка
На самом деле уравнение Михаэлиса-Ментен в данном виде предложили Бриггс и Холдейн, но в честь основоположников оно носит название Михаэлиса-Ментен.
Выделяют три основных решения уравнения Михаэлиса-Ментен:
1. Концентрация субстрата равна величине константы Михаэлиса ([S] = Km).
В этом случае, решая уравнение Михаэлиса-Ментен, получаем, что скорость реакции V будет равна половине максимальной Vmax.(V = ½ Vmax).
В математическом смысле Km соответствует концентрации субстрата при которой скорость реакции равна половине максимальной. Ее биологический смысл заключается в характеристике сродства фермента к субстрату, а именно: увеличение величины Кm означает снижение сродства фермента к субстрату.
2. Концентрация субстрата значительно больше Km ([S] >> Km). В этом случае величиной Km можно пренебречь, при решении получим, что скорость реакции максимальна (плато на графике).
3. Концентрация субстрата значительно меньше Km ([S]
Кинетика ферментативных реакций
Одним из характерных проявлений жизни является удивительная способность живых организмов кинетически регулировать химические реакции, подавляя стремление к достижению термодинамического равновесия. Ферментативная кинетика занимается исследованием закономерностей влияния химической природы реагирующих веществ (ферментов, субстратов) и условий их взаимодействия (концентрация, рН среды, температуры, присутствие активаторов или ингибиторов) на скорость ферментативной реакции. Главной целью изучения кинетики ферментативных реакций является получение информации, которая может способствовать выяснению молекулярного механизма действия фермента.
Общие принципы кинетики химических реакций применимы и к ферментативным реакциям. Известно, что любая химическая реакция характеризуется константой термодинамического равновесия. Она выражает состояние химического равновесия, достигаемого системой, и обозначается Кр. Так, для реакции:
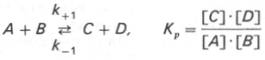
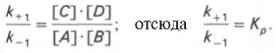
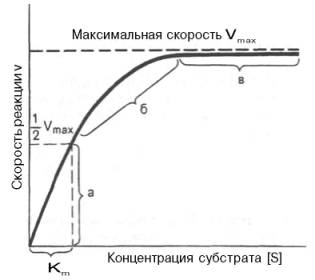
Рис. 4.12. Теоретический график зависимости скорости ферментативной реакции от концентрации субстрата при постоянной концентрации фермента.
а — реакция первого порядка (при [ S ]
Таким образом, константа равновесия равна отношению констант скоростей прямой и обратной реакций. Величину, обратную константе равновесия, принято называть субстратной константой, или, в случае ферментативной реакции, константой диссоциации фермент–субстратного комплекса, и обозначать символом KS. Так, в реакции
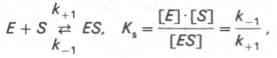
т.е. KSравна отношению произведения концентрации фермента и субстрата к концентрации фермент-субстратного комплекса или отношению констант скоростей обратной и прямой реакций. Следует отметить, что константа KSзависит от химической природы субстрата и фермента и определяет степень их сродства. Чем ниже значение KS, тем выше сродство фермента к субстрату.
При изучении кинетики ферментативных реакций следует учитывать одну важную особенность этих реакций (не свойственную обычным химическим реакциям), связанную с явлением насыщения фермента субстратом. При низкой концентрации субстрата зависимость скорости реакции от концентрации субстрата (рис. 4.12) является почти линейной и подчиняется кинетике первого порядка. Это означает, что скорость реакции S —> Р прямо пропорциональна концентрации субстрата S и в любой момент времени t определяется следующим кинетическим уравнением:
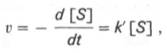
где [S] – молярная концентрация субстрата S; –d[S]/dt – скорость убыли субстрата; k’ – константа скорости реакции, которая в данном случае имеет размерность, обратную единице времени (мин –1 или с –1 ).
При высокой концентрации субстрата скорость реакции максимальна, становится постоянной и не зависящей от концентрации субстрата [ S ] . В этом случае реакция подчиняется кинетике нулевого порядка v = k» (при полном насыщении фермента субстратом) и целиком определяется концентрацией фермента. Различают, кроме того, реакции второго порядка, скорость которых пропорциональна произведению концентраций двух реагирующих веществ. В определенных условиях при нарушении пропорциональности говорят иногда о реакциях смешанного порядка (см. рис. 4.12).
Изучая явление насыщения, Л. Михаэлис и М. Ментен разработали общую теорию ферментативной кинетики. Они исходили из предположения, что ферментативный процесс протекает в виде следующей химической реакции:
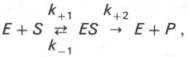
т.е. фермент Е вступает во взаимодействие с субстратом S с образованием промежуточного комплекса ES, который далее распадается на свободный фермент и продукт реакции Р. Математическая обработка на основе закона действующих масс дала возможность вывести уравнение, названное в честь авторов уравнением Михаэлиса–Ментен, выражающее количественное соотношение между концентрацией субстрата и скоростью ферментативной реакции:
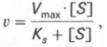
Из уравнения Михаэлиса–Ментен следует, что при высокой концентрации субстрата и низком значении KSскорость реакции является максимальной, т.е. v = Vmax(реакция нулевого порядка, см. рис. 4.12). При низкой концентрации субстрата, напротив, скорость реакции оказывается пропорциональной концентрации субстрата в каждый данный момент (реакция первого порядка).
Следует указать, что уравнение Михаэлиса–Ментен в его классическом виде не учитывает влияние на скорость ферментативного процесса продуктов реакции, например в реакции
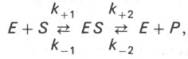
и носит несколько ограниченный характер. Поэтому были предприняты попытки усовершенствовать его. Так, было предложено уравнение Бриггса-Холдейна:
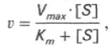
где Кm представляет собой константу Михаэлиса, являющуюся экспериментально определяемой величиной. Она может быть представлена следующим уравнением:
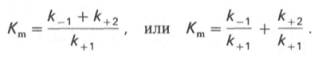
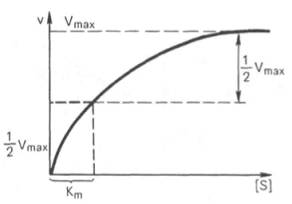
Рис. 4.13. Кривая уравнения Михаэли-са-Ментен: гиперболическая зависимость начальных скоростей катализируемой ферментом реакции от концентрации субстрата.
В числителе представлены константы скоростей распада комплекса ES в двух направлениях (в сторону исходных Е и S и в сторону конечных продуктов реакции Е и Р). Отношение k–1/ k+1представляет собой константу диссоциации ферментсубстратного комплекса KS, тогда:
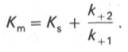
Отсюда вытекает важное следствие: константа Михаэлиса всегда больше константы диссоциации фермент-субстратного комплекса KSна величину
Для определения численного значения Кm обычно находят ту концентрацию субстрата, при которой скорость ферментативной реакции v составляет половину от максимальной Vmax, т.е. если v = 1 /2 Vmaх. Подставляя значение v в уравнение Бриггса–Холдейна, получаем:
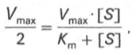
разделив обе части уравнения на Vmах, получим
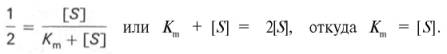
Таким образом, константа Михаэлиса численно равна концентрации субстрата (моль/л), при которой скорость данной ферментативной реакции составляет половину от максимальной.
Определение величины Кm имеет важное значение при выяснении механизма действия эффекторов на активность ферментов и т.д. Константу Михаэлиса можно вычислить по графику (рис. 4.13). Отрезок на абсциссе, соответствующий скорости, равной половине максимальной, будет представлять собой Кm.
Пользоваться графиком, построенным в прямых координатах зависимости начальной скорости реакции v0 от начальной концентрации субстрата [S0], неудобно, поскольку максимальная скорость Vmaxявляется в данном случае асимптотической величиной и определяется недостаточно точно.
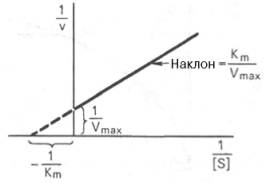
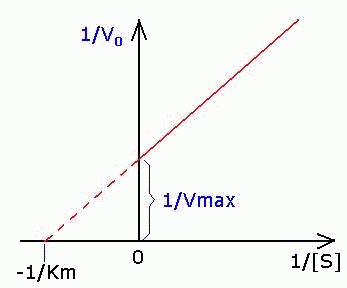
Рис. 4.14. График Лайнуивера-Бэрка.
Для более удобного графического представления экспериментальных данных Г. Лайнуивер и Д. Бэрк преобразовали уравнение Бриггса–Хол-дейна по методу двойных обратных величин исходя из того принципа, что если существует равенство между двумя какими-либо величинами, то и обратные величины также будут равны. В частности, если
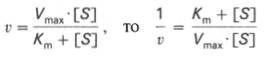
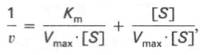
то после преобразования получаем уравнение:
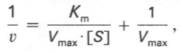
которое получило название уравнения Лайнуивера–Бэрка. Это уравнение прямой линии: у = ах + b. Если теперь в соответствии с этим уравнением построить график в координатах 1/v (y) от l/[S] (x), то получим прямую линию (рис. 4.14), тангенс угла наклона который будет равен величине Km/Vmax; отрезок, отсекаемый прямой от оси ординат, представляет собой l/Vmax(обратная величина максимальной скорости). Если продолжить прямую линию за ось ординат, тогда на абсциссе отсекается отрезок, соответствующий обратной величине константы Михаэлиса – 1/Кm (см. рис. 4.14). Таким образом, величину Кm можно вычислить из данных наклона прямой и длины отрезка, отсекаемого от оси ординат, или из длины отрезка, отсекаемого от оси абсцисс в области отрицательных значений.
Следует подчеркнуть, что значения Vmax, как и величину Кm, более точно, чем по графику, построенному в прямых координатах, можно определить по графику, построенному по методу двойных обратных величин. Поэтому данный метод нашел широкое применение в современной энзимологии. Предложены также аналогичные графические способы определения Кm и Vmaxв координатах зависимости v от v/[S] и [S]/v от [S].
Следует отметить некоторые ограничения применения уравнения Ми-хаэлиса–Ментен, обусловленные множественными формами ферментов и аллостерической природой фермента. В этом случае график зависимости начальной скорости реакции от концентрации субстрата (кинетическая
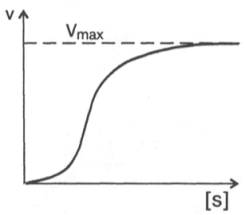
Рис. 4.15. Сигмоидная кинетическая кривая насыщения субстратом.
кривая) имеет не гиперболическую форму, а сигмоидный характер (рис. 4.15) наподобие кривой насыщения гемоглобина кислородом. Это означает, что связывание одной молекулы субстрата в одном каталитическом центре повышает связывание субстрата с другим центром, т.е. имеет место кооперативное взаимодействие, как и в случае присоединения кислорода к 4 субъединицам гемоглобина. Для оценки концентрации субстрата, при которой скорость реакции составляет половину максимальной, в условиях сигмоидного характера кинетической кривой обычно применяют преобразованное уравнение Хилла:
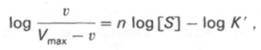
где К’ – константа ассоциации; n – число субстратсвязывающих центров.
Кинетика ферментативных процессов уравнение михаэлиса ментен
МЕХАНИЗМ ФЕРМЕНТАТИВНОГО КАТАЛИЗА
Катализ приводит к ускорению достижения равновесия за счет снижения энергии активации (Еа), часто ступенчато.
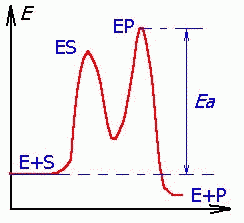 | Три стадии процесса: 1) E + S —— ES (K = k1/k-1) (БЫСТРАЯ) 2) ES —— EP (k2)(медленная) 3) EP —- E + P Таким образом, в момент равновесия скорости образования и исчезновения энзимсубстратного комплекса (ES) равны: E + S —- ES —— EP — E + P |
ВЛИЯНИЕ НА НАЧАЛЬНУЮ СКОРОСТЬ РЕАКЦИИ КОНЦЕНТРАЦИЙ ФЕРМЕНТА [E] и СУБСТРАТА [S].
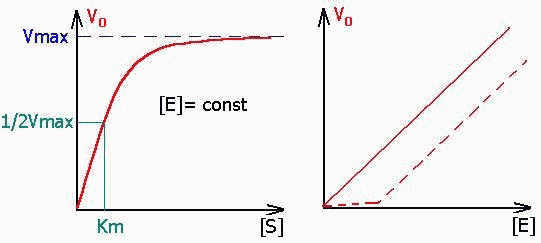 | [E]- ЗАВ-ТЬ ЛИНЕЙНАЯ, [S]- ЗАВ-ТЬ ГИПЕРБОЛИЧЕСКАЯ. (Это значит, что при малых S- порядок первый, далее переходит в нулевой). Пунктиром на графике зависимости скорости от концентрации фермента показано влияние ферментных ядов (ингибиторов), проявляющееся в медленном нарастании скорости реакции до момента, пока не будет преодолено влияние яда повышением количества энзима. |
УРАВНЕНИЕ МИХАЭЛИСА — МЕНТЕН
При формулировке кинетического выражения для скорости ферментативной реакции Бриггс и Холдейн (Briggs & Haldane, 1925) сделали три допущения:
1) Стационарное состояние реакции в момент равновесия, когда скорости образования и расходования ES равны;
2) Весь фермент в условиях насыщающих концентраций субстрата превращается в энзимсубстратный комплекс ES;
3) Если весь фермент в виде ES, то скорость реакции максимальна и Vmax=k2[ES].
Образование ES: [ES]=k1[S][E] (I)
Расходование ES: [ES]=k-1[ES]+k2[ES] (II)
Приравнивая выражения (I) и (II) и сокращая обе части на k1 получаем:
[S][E] = [ES](k-1 + k2)/k1 = [ES]Km, где Km = (k-1 + k2)/k1
Выразим равновесную концентрацию [E] через начальную [Eo]: [E] = [Eo] — [ES]
[S]([Eo]-[ES])= [ES]Km, переносим [S] в правую часть выражения и делим обе части на [ES]:
[Eo]/[ES]=Km/[S]+1= (Km+[S])/[S] (III)
Поскольку трудно (если не невозможно) измерить [ES], произведем замену с учетом того, что в насыщающих концентрациях [S] весь [Eo] перейдет в [ES] и максимальная скорость при этом будет равна Vmax=k2[ES]=k2[Eo].
В это же время скорость реакции равна V=k2[ES]. Через отношение этих скоростей выразим [Eo]/[ES]:
V/Vmax= [ES]/[Eo]
В уравнении (III) произведем замену отношения [Eo]/[ES] на Vmax/V и получаем:
V = Vmax[S]/(Km+[S])
Это и есть уравнение Михаэлиса-Ментен. Название было закреплено за ними, несмотря на то, что Леонор Михаэлис и Мод Ментен только постулировали положение о том, что фермент обратимо связывается с субстратом (1913). Основу ферментативной кинетики положил Виктор Генри в 1903 году, впервые предположивший зависимость между концентрациями субстрата и фермента в ферментативных реакциях.
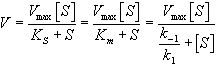
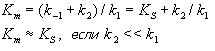
Графическая зависимость для уравнения имеет вид:
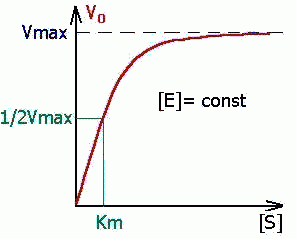 | Константа Михаэлиса измеряется в молях на литр и бывает от 10 -2 до 10 -7 , чем меньше Кm, тем активнее фермент. При V=1/2Vmax, имеем Km = [S]. Однако, определение Vmax затруднительно по асимптоте. Для устранения этого неудобства ХАНС ЛАЙНУИВЕР и ДИН БЭРК приравняли обратные зависимости левой и правой частей уравнения. |
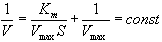
Графическое выражение для скорости реакции в координатах Лайнуивера-Бэрка имеет вид прямой линии, отсекающей на оси Х значение -1/Km, а на оси Y- значение 1/V max:
ВЛИЯНИЕ ТЕМПЕРАТУРЫ И рН НА АКТИВНОСТЬ ФЕРМЕНТА
В общем, для ферментов имеется связь константы скорости и температуры, выражаемая: 2,3 lgK = B- Ea/RT, где Еа- энергия активации, В-частота столкновений.
Однако, указанная зависимость прямолинейна лишь на ограниченном отрезке, так как с ростом температуры наблюдается денатурация фермента.
Зависимость активности фермента от рН также проходит через максимум. Кислотность среды влияет на заряды в молекуле фермента и, тем самым, на его способность к связыванию субстрата.
Единицы активности:
Е (станд. ед) — количество Ф., которое превращает 1 мкМоль субстрата за 1 мин.
КАТАЛ- кол-во Ф., превращающее субстрат со скоростью 1 Моль/сек.
1 КАТАЛ = 60 000 000 Е.
2) НЕОБРАТИМЫЕ (КАК ПРАВИЛО- НЕПРИРОДНОГО ПРОИСХОЖДЕНИЯ — NaF (ингибитор фосфатаз), п-хлормеркурибензоат (связывает группировки -SH), диизопропилфторфосфат (протеиназ и эстераз)- эти ингибиторы действуют необратимо.
ОБРАТИМЫЕ ИНГИБИТОРЫ- клеточные метаболиты и типы ингибирования
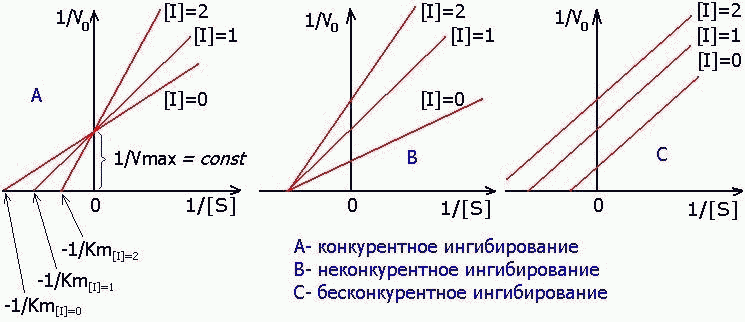
а) КОНКУРЕНТНОЕ ингибирование
б) НЕКОНКУРЕНТНОЕ ингибирование
в) БЕСКОНКУРЕНТНОЕ ингибирование
а) КОНКУРЕНТНОЕ ИНГИБИРОВАНИЕ- когда за активный центр фермента вместе с субстратом конкурирует ИНГИБИТОР:
Е + I = EI Ki = [E][I] / [EI], чем меньше Ki, тем сильнее ингибитор.
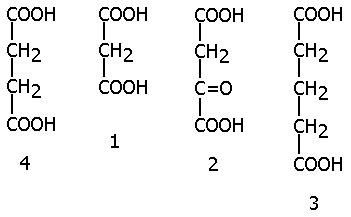 | Так, малоновая (1), щавелевоуксусная (2) и глутаровая (3) кислоты ингибируют фермент СУКЦИНАТДЕГИДРОГЕНАЗУ, субстратом которой является янтарная кислота (4) (СУКЦИНАТ), так как они сходны по строению с субстратом: |
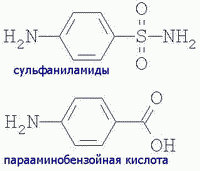 | Другой пример. Сульфаниламидные антибактериальные препараты имеют сходное строение с парааминобензойной кислотой и являются конкурентными ингибиторами в синтезе бактериями фолиевой кислоты (фактора роста бактерий). У человека нет такого метаболического пути и в лечебных дозах они не влияют на жизнедеятельность человека, оказывая общий БАКТЕРИОСТАТИЧЕСКИЙ эффект (нарушая в некоторой степени деятельность кишечной микрофлоры): |
2) НЕКОНКУРЕНТНОЕ ИНГИБИРОВАНИЕ ПРОЯВЛЯЕТСЯ ПРИ СВЯЗЫВАНИИ ИНГИБИТОРА С ФЕРМЕНТОМ ВНЕ АКТИВНОГО ЦЕНТРА, но при этом меняется структура активного центра и связь с субстратом становится невозможной.
E + I —> EI , EI + S —-> (невозможно)
3) БЕСКОНКУРЕНТНОЕ ИНГИБИРОВАНИЕ — результат присоединения ингибитора ТОЛЬКО ПОСЛЕ образования ЭНЗИМСУБСТРАТНОГО КОМПЛЕКСА:
E + S = ES ES + I = ESI
ОПРЕДЕЛЕНИЕ ТИПА ИНГИБИРОВАНИЯ
1) КОНКУРЕНТНЫЙ ИНГИБИТОР увеличивает Кm и не изменяет V max.
2) НЕКОНКУРЕНТНЫЙ ингибитор не изменяет Кm и снижает V max.
3) БЕСКОНКУРЕНТНЫЙ ингибитор в одинаковой степени снижает Кm и V max:
Некоторые продукты ферментативных реакций также выступают в роли ингибиторов. Так, глюкоза ингибирует фермент Г-6-фосфатазу:
Глюкозо-6-фосфат + Н 2 О — Глюкоза + Н 3 РО 4
Ингибирование избытком субстрата наблюдается в ряде случаев в результате блокирования активного центра по схеме:
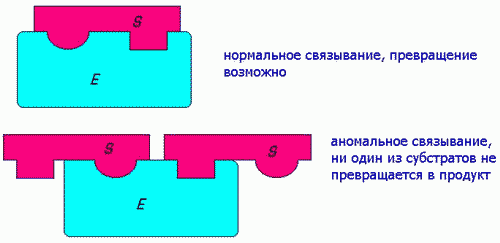
1) ПУТЕМ ЗАЩИТЫ Ф. ОТ ИНАКТИВИР. ВЛИЯНИЙ
2) ПУТЕМ ВОЗДЕЙСТВИЯ НА СУБЪЕДИНИЦЫ БЕЛКОВ Ф.
Ферменты-протеинкиназы имеют 4-ную структуру, состоят их 2-х субъед- каталитической и регуляторной. Активатор воздействует на регуляторную, а в результате обнажается каталитическая субъединица:
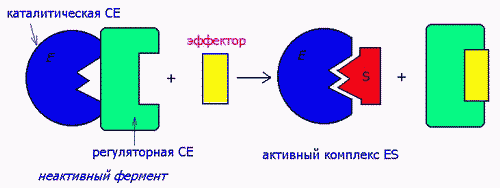
Аллостерическими эффекторами называют ингибиторы (отрицательные эффекторы) и активаторы (положительные эффекторы) энзимов, регулирующие активность ферментов в широких пределах, не затрагивая при этом активного центра, поскольку воздействуют на него опосредованно, присоединясь к молекуле фермента ВНЕ активного центра. Они способны управлять конформацией активного центра таким образом, что присоединение субстрата либо облегчается значительно, либо вовсе становится невозможным:
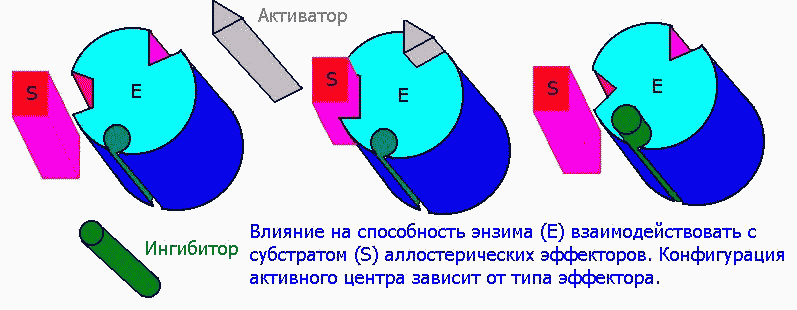
1) Структура аллостерических эффекторов отлична от природного субстрата фермента.
2) Высокоспецифичны
3) Катализируют фермент первой цепи процесса.
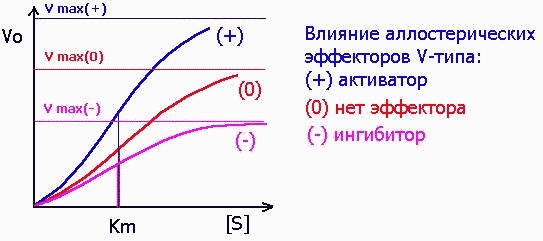
Среди аллостерических эффекторов различают эффекторы V-типа, которые подобно неконкурентным ингибиторам (см. выше) изменяют значение Vmax:
и эффекторы К-типа, изменяющие значение Km, как это свойственно конкурентным ингибиторам: 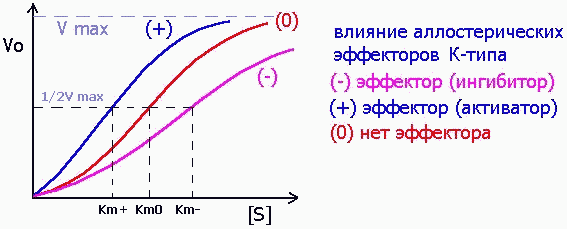
Для ферментов с аллостерической регуляцией, а также и для ферментов с четвертичной структурой, состоящих из нескольких субединиц, характерны сигмоидальные (S-образные) кинетические кривые.
КООПЕРАТИВНЫЙ ЭФФЕКТ
Характерен для ферментов, имеющих 2 и более субъединиц. Присоединение субстрата или эффектора к одной из субъединиц облегчает последующие присоединения к оставшимся субъединицам. Типичный пример кооперативного эффекта- перенос кислорода молекулами гемоглобина.
ПРОФЕРМЕНТЫ
Неактивная форма Ф. Активация осуществляется удалением части полипептидной цепи, обнажению активного центра, при помощи специфических ферментов. Биологический смысл- в месте продукции Ф. мог бы вызвать нежелательные последствия (переваривание и т.д.).
ИЗОФЕРМЕНТЫ
отличные по структуре, но выполняющие одну и ту же функцию. Изоферментами, в частности, существуют алкогольдегидрогеназа и целых 5 изомеров лактатдегидрогеназы. Изоферменты могут различаться по каталитической активности, несмотря на то, что катализируют одну и ту же реакцию. Так, в клетках печени человека различаются цитоплазматическая ацетальдегиддегидрогеназа (низкоактивная, с высоким значением Km) и митохондриальная ацетальдегиддегидрогеназа (AcDH), с малым значением Km. Последнее обстоятельство является биохимической причиной непереносимости алкоголя у коренного населения Юго-Восточной Азии, которое связано с практическим отсутствием у них (по генетическим причинам) митохондриальной AcDH и превращение образовавшегося из этанола ацетальдегида осуществляется у них малоактивной цитоплазматической AcDH.
ПРИМЕНЕНИЕ ФЕРМЕНТОВ В ДИАГНОСТИКЕ И ДРУГИХ ОБЛАСТЯХ
Ферментные препараты и регуляторы их активности находят все более широкое применение в различных областях медицины, промышленности и науки.
В медицине используются энизимы, как терапевтические средства:
— СТРЕПТОКИНАЗА- смесь энзимов из семейства Streptococcus, активизирующих ПЛАЗМИНОГЕН и образование ПЛАЗМИНА, приводящее к растворению ФИБРОЗНЫХ ТРОМБОВ в кровеносных сосудах. Рекомбинантный тканевый активатор плазминогена rh-tPA производится генно-инженерным способом на E.Coli и вводится человеку.
— АСПАРАГИНАЗА используется в терапии ЛЕЙКЕМИИ взрослых. Опухолевые клетки нуждаются в АСПАРАГИНЕ, который они извлекают из плазмы крови. Внутривенное введение (i.v.) аспарагиназы снижает содержание аспарагина и замедляет рост раковых клеток. К сожалению, достижение высоких терапевтических доз таким способом невозможно.
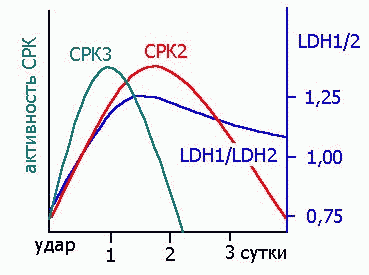
ЭНЗИМЫ, КАК ДИАГНОСТИЧЕСКИЕ ПОКАЗАТЕЛИ
 |