ВАНТ-ГОФФА ПРАВИЛО
ВАНТ-ГОФФА ПРАВИЛО. Почти все химические реакции при повышении температуры идут быстрее. Зависимость скорости реакции от температуры описывается уравнением Аррениуса:
k = Ae –E a/RT, где k – константа скорости реакции, А – не зависящая от температуры константа (ее называют предэкспоненциальным множителем), Еа– энергия активации, R – газовая постоянная, Т – абсолютная температура. В школьных учебниках зависимость скорости реакции от температуры определяют в соответствии с так называемым «правилом Вант-Гоффа», которое в 19 в. сформулировал голландский химик Якоб Вант-Гофф. Это чисто эмпирическое правило, т.е. правило, основанное не на теории, а выведенное из опытных данных. В соответствии с этим правилом, повышение температуры на 10° приводит к увеличению скорости в 2–4 раза. Математически эту зависимость можно выразить уравнением v2v1 = g (T2 – T 1 )/10 , где v1 и v2– скорости реакции при температурах Т1 и Т2; величина g называется температурным коэффициентом реакции. Например, если g = 2, то при Т2– Т1 = 50 о v2/v1 = 2 5 = 32, т.е. реакция ускорилась в 32 раза, причем это ускорение никак не зависит от абсолютных величин Т1 и Т2, а только от их разности.
Однако из уравнения Аррениуса следует, что температурный коэффициент реакции зависит как от энергии активации, так и от абсолютной температуры. Для данной реакции с определенным значением Еа ускорение при повышении температуры на 10° будет тем больше, чем ниже температура. Это почти очевидно и без расчетов: повышение температуры от 0 до 10° С должно сказаться на скорости реакции значительно сильнее, чем такое же повышение температуры, например, от 500 до 510° С.
С другой стороны, для данного температурного интервала ускорение реакции будет тем сильнее, чем больше ее энергия активации. Так, если энергия активации реакции мала, то такая реакция идет очень быстро, и при повышении температуры на 10° С ее скорость почти не изменяется. Для таких реакций температурный коэффициент намного меньше 2. Для реакций же с большой энергией активации, которые при невысоких температурах идут медленно, ускорение при повышении температуры на 10° С может значительно превысить 4-кратное.
Например, реакция диоксида углерода со щелочным раствором с образованием гидрокарбонат-иона (СО2 + ОН® НСО3 – ) имеет энергию активации 38,2 кДж/моль, поэтому при повышении температуры, например, от 50 до 60° С эта реакция ускорится всего в 1,5 раза. В то же время реакция распада этилбромида на этилен и бромоводород (С2Н5Вr ® С2Н4 + НВr) с энергией активации 218 кДж/моль ускорится при повышении температуры от 100 до 110 o С в 6,3 раза (правда, в этом интервале температур реакция идет очень медленно). Кинетика реакции атомов водорода с этаном H + C2H6 ® H2 + C2H5 была изучены в широком температурном интервале – от 300 до 1100 К (27–827° С). Для этой реакции Eа = 40,6 кДж/моль. Следовательно, повышение температуры на 10° вызовет увеличение скорости реакции в 1,7 раза в интервале 300–310 K и только в 1,04 раза в интервале 1090–1100 K. Так что при высоких температурах скорость этой реакции практически не зависит от температуры. А для реакции присоединения атома водорода к двойной связи H + C2H4 ® C2H5 энергия активации мала (Eа = 3,4 кДж/моль, так что ее скорость слабо зависит от температуры в широком температурном интервале. И только при температурах намного ниже 0° С начинает сказываться наличие активационного барьера.
Подобных примеров можно привести множество. Очевидно, что правило Вант-Гоффа противоречит не только уравнению Аррениуса, но и многим экспериментальным данным. Откуда же оно взялось и почему нередко выполняется?
Если в приведенном выше математическом выражении для правила Вант-Гоффа подставить вместо скоростей v1 и v2 для данной реакции их зависимости от температуры, в соответствии с уравнением Аррениуса, то после сокращения предэкспоненциальных множителей получим следующее выражение: g = vT +10/vT = е –Е а/R(Т+10)/е –Е а/RТ = е (Еа/R)[1/Т – 1/(T+10)] . Логарифмироване этого уравнения дает: lng = (Eа/R)[1/T – 1/(T + 10)], откуда Еа = Rlng T(T + 10)/10 = 0,83lngT(T + 10). Энергия активации не является функцией температуры, эта зависимость нужна лишь для удобства последующего анализа. Последнее уравнение – это уравнение параболы, в котором физический смысл имеют только положительные значения. Соответствующая диаграмма ограничена двумя ветвями параболы: при g = 2 получаем Еа = 0,58Т(Т + 10), при g = 4 получаем Еа = 1,16Т(Т + 10). К тем же формулам приходим и при использовании десятичных логарифмов. Соответствующие графики двух парабол, для значений g 2 и 4, приведены на рисунке. Их физический смысл заключается в том, что области выполнения правила Вант-Гоффа соответствует только область между параболами. Таким образом, существуют только определенные соотношения между энергией активации реакции и температурой ее проведения, при которых правило Вант-Гоффа выполняется. Ниже нижней ветви температурный коэффициент g 4.
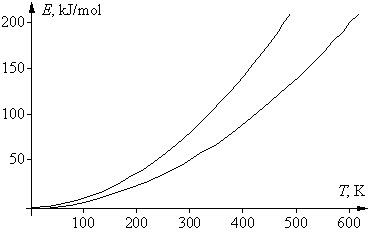
Если посмотреть, какие реакции «укладываются» в указанную довольно узкую область, то окажется, что все эти реакции идут не слишком быстро и не слишком медленно, а с удобной для измерения (при данной температуре) скоростью. Скорость только таких реакций и могли изучать химики во времена Вант-Гоффа. Например, если энергия активации была невелика (меньше 50 кДж/моль), то такая реакция при комнатной температуре заканчивалась за 1–2 секунды; поэтому для изучения ее кинетики следовало значительно понизить температуру, чтобы реакция проходила не быстрее, чем за 10–20 минут. Только в этом случае химики 19 в. успевали отбирать пробы по ходу реакции и анализировать изменение в них концентрации реагентов. Других способов изучения скорости реакции в то время не было. Если это не удавалось (например, водный раствор замерзал), то скорость такой реакции не изучали. Если же энергия активации реакции была велика и при комнатной температуре она шла слишком медленно (многие сутки, или даже недели), то температуру повышали, чтобы реакция шла с удобной для измерения скоростью. И здесь были свои ограничения – например, раствор мог закипеть, т.е. в любом случае исследователи фактически «подстраивали» изучаемую реакцию под область между двумя параболами.
Сейчас химики имеют возможность с помощью различных приборов экспериментально изучать и очень быстрые (идущие в микросекундной области), и очень медленные реакции, для которых температурный коэффициент может быть значительно меньше 2 или значительно больше 4. Поэтому правило Вант-Гоффа, которое, в отличие от уравнения Аррениуса, не имеет четкого физического смысла, представляет лишь чисто исторический интерес и в современной науке не используется. В подавляющем большинстве учебников и монографий по химической кинетике, а также в 5-томной Химической Энциклопедии это правило даже не упоминается. И, тем не менее, если изучаемая реакция идет с удобной для измерения скоростью, например, заканчивается за 30–40 мин, а энергия активации ее еще не измерена, то для предварительной грубой оценки зависимости скорости такой реакции от температуры можно использовать правило Вант-Гоффа. Поэтому это правило приводится во всех школьных учебниках химии.
Коллигативные свойства растворов
Любому раствору характерны те или иные физические свойства, к которым относятся и коллигативные свойства растворов. Это такие свойства, на которые не оказывает влияние природа растворенного вещества, а зависят они исключительно от количества частиц этого растворенного вещества.
К коллигативным свойствам растворов относятся:
- Понижение давление паров
- Повышение температуры кипения
- Понижение температуры затвердевания (кристаллизации)
- Осмотическое давление раствора.
Рассмотрим подробнее каждое из перечисленных свойств.
Понижение давления паров
Давление насыщенного пара (т.е. пара, который пребывает в состоянии равновесия с жидкостью) над чистым растворителем называется давлением или упругостью насыщенного пара чистого растворителя.
Если в некотором растворителе растворить нелетучее вещество, то равновесное давление паров растворителя при этом понижается, т.к. присутствие какого – либо вещества, растворенного в этом растворителе, затрудняет переход частиц растворителя в паровую фазу.
Экспериментально доказано, что такое понижение давления паров напрямую зависит от количества растворенного вещества. В 1887 г. Ф.М. Рауль описал количественные закономерности коллигативных свойств растворов.
Первый закон Рауля
Первый закон Рауля заключается в следующем:
Давление пара раствора, содержащего нелетучее растворенное вещество, прямо пропорционально мольной доле растворителя в данном растворе:
p — давление пара над раствором, Па;
p0 — давление пара над чистым растворителем, Па;
χр-ль — мольная доля растворителя.
nв-ва и nр-ля – соответственно количество растворенного вещества и растворителя, моль.
Иногда Первому закону Рауля дают другую формулировку:
относительное понижение давления насыщенного пара растворителя над раствором равно мольной доле растворенного вещества:
При этом принимаем, что χв-ва + χр-ль= 1
Изотонический коэффициент Вант-Гоффа
Для растворов электролитов данное уравнение приобретает несколько иной вид, в его состав входит изотонический коэффициент i:
Δp — изменение давления паров раствора по сравнению с чистым растворителем;
i – изотонический коэффициент.
Изотонический коэффициент (или фактор Вант-Гоффа) — это параметр, не имеющий размерности, который характеризует поведение какого – либо вещества в растворе.
То есть, изотонический коэффициент показывает, разницу содержания частиц в растворе электролита по сравнению с раствором неэлектролита такой же концентрации. Он тесно связан связан с процессом диссоциации, точнее, со степенью диссоциации и выражается следующим выражением:
n – количество ионов, на которые диссоциирует вещество.
α – степень диссоциации.
Повышение температуры кипения или понижение температуры затвердевания (кристаллизации). Второй закон Рауля
Равновесное давление паров жидкости имеет тенденцию к увеличению с ростом температуры, жидкость начинает кипеть, при уравнивании давления ее паров и внешнего давления.
При наличии нелетучего вещества, давление паров раствора снижается, и раствор будет закипать при более высокой температуре, по сравнению с температурой кипения чистого растворителя.
Температура замерзания жидкости также определяется той температурой, при которой давления паров жидкой и твердой фаз уравниваются.
Ф.М. Рауль доказал, что повышение температуры кипения, так же как и понижение температуры замерзания разбавленных растворов нелетучих веществ, прямо пропорционально моляльной концентрации раствора и не зависит от природы растворённого вещества. Это правило известно как Второй закон Рауля:
K — криоскопическая константа,
mв-ва — моляльность вещества в растворе.
Растворы электролитов не подчиняются Законам Рауля. Но для учёта всех несоответствий Вант-Гофф предложил ввести в приведённые уравнения поправку в виде изотонического коэффициента i, учитывающего процесс распада на ионы молекул растворённого вещества:
Осмотическое давление раствора
Некоторые материалы имеют способность к полупроницаемости, т.е. им свойственно пропускать частицы определенного вида и не пропускать частицы другого вида.
Перемещение молекул растворителя (но не растворенного, в нем вещества), через полупроницаемую мембрану в раствор с большей концентрацией из более разбавленного представляет собой такое явление как осмос.
Представим два таких раствора, которые разделены полупроницаемой мембраной, как показано на рисунке выше. Растворы стремятся к выравниванию концентраций, поэтому вода будет проникать в раствор, тем самым уменьшая его концентрацию.
Для того, чтобы осмос приостановить, необходимо приложить внешнее давление к раствору. Такое давление, которое требуется приложить, называется осмотическим давлением.
Осмотическое давление и концентрацию раствора неэлектролита позволяет связать уравнение Вант — Гоффа, которое напоминает уравнение идеального газа Клапейрона – Менделеева:
где C — молярная концентрация раствора, моль/м 3 ,
R — универсальная газовая постоянная (8,314 Дж/моль·К);
T — абсолютная температура раствора.
Преобразуем уравнение следующим образом:
C = n/V = m/(M·V)
π = т·R·T / M·V или
Для растворов электролитов осмотическое давление определяется уравнением, в которое входит изотонический коэффициент:
где i — изотонический коэффициент раствора.
Для растворов электролитов i > 1, а для растворов неэлектролитов i = 1.
Если полупроницаемой перегородкой разделены два раствора, имеющие одинаковое осмотическое давление, то перемещение растворителя через перегородку отсутствует. Такие растворы называются изотоническими.
Раствор, с меньшим осмотическим давлением, по сравнению с более концентрированным раствором, называют гипотоническим, а раствор с большей концентрацией – гипертоническим.
Химическая кинетика. Скорость химических реакций
Темы кодификатора ЕГЭ: Скорость реакции. Ее зависимость от разных факторов.
Скорость химической реакции показывает, как быстро происходит та или иная реакция. Взаимодействие происходит при столкновении частиц в пространстве. При этом реакция происходит не при каждом столкновении, а только когда частица обладают соответствующей энергией.
Скорость реакции – количество элементарных соударений взаимодействующих частиц, заканчивающихся химическим превращением, за единицу времени.
Определение скорости химической реакции связано с условиями ее проведения. Если реакция гомогенная – т.е. продукты и реагенты находятся в одной фазе – то скорость химической реакции определяется, как изменение концентрации вещества в единицу времени:
υ = ΔC / Δt
Если реагенты, или продукты находятся в разных фазах, и столкновение частиц происходит только на границе раздела фаз, то реакция называется гетерогенной, и скорость ее определяется изменением количества вещества в единицу времени на единицу реакционной поверхности:
υ = Δν / (S·Δt)
Факторы, влияющие на скорость химической реакции
1. Температура
Самый простой способ изменить скорость реакции – изменить температуру . Как вам, должно быть, известно из курса физики, температура – это мера средней кинетической энергии движения частиц вещества. Если мы повышаем температуру, то частицы любого вещества начинают двигаться быстрее, а следовательно, сталкиваться чаще.
Однако при повышении температуры скорость химических реакций увеличивается в основном благодаря тому, что увеличивается число эффективных соударений. При повышении температуры резко увеличивается число активных частиц, которые могут преодолеть энергетический барьер реакции. Если понижаем температуру – частицы начинают двигаться медленнее, число активных частиц уменьшается, и количество эффективных соударений в секунду уменьшается. Таким образом, при повышении температуры скорость химической реакции повышается, а при понижении температуры — уменьшается .
Обратите внимание! Это правило работает одинаково для всех химических реакций (в том числе для экзотермических и эндотермических). Скорость реакции не зависит от теплового эффекта. Скорость экзотермических реакций при повышении температуры возрастает, а при понижении температуры – уменьшается. Скорость эндотермических реакций также возрастает при повышении температуры, и уменьшается при понижении температуры.
Более того, еще в XIX веке голландский физик Вант-Гофф экспериментально установил, что скорость большинства реакций примерно одинаково изменяется (примерно в 2-4 раза) при изменении температуры на 10 о С.
Правило Вант-Гоффа звучит так: повышение температуры на 10 о С приводит к увеличению скорости химической реакции в 2-4 раза (эту величину называют температурный коэффициент скорости химической реакции γ).
Точное значение температурного коэффициента определяется для каждой реакции.

здесь v2 — скорость реакции при температуре T2,
v1 — скорость реакции при температуре T1,
γ — температурный коэффициент скорости реакции, коэффициент Вант-Гоффа.
В некоторых ситуациях повысить скорость реакции с помощью температуры не всегда удается, т.к. некоторые вещества разлагаются при повышении температуры, некоторые вещества или растворители испаряются при повышенной температуре, т.е. нарушаются условия проведения процесса.
2. Концентрация
Также изменить число эффективных соударений можно, изменив концентрацию реагирующих веществ . Понятие концентрации, как правило, используется для газов и жидкостей, т.к. в газах и жидкостях частицы быстро двигаются и активно перемешиваются. Чем больше концентрация реагирующих веществ (жидкостей, газов), тем больше число эффективных соударений, и тем выше скорость химической реакции.
На основании большого числа экспериментов в 1867 году в работах норвежских ученых П. Гульденберга и П. Вааге и, независимо от них, в 1865 году русским ученым Н.И. Бекетовым был выведен основной закон химической кинетики, устанавливающий зависимость скорости химической реакции от концентрации реагирующих веществ:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ в степенях, равных их коэффициентам в уравнении химической реакции.
Для химической реакции вида: aA + bB = cC + dD закон действующих масс записывается так:

здесь v — скорость химической реакции,
CA и CB — концентрации веществ А и В, соответственно, моль/л
k – коэффициент пропорциональности, константа скорости реакции.
Например , для реакции образования аммиака:
закон действующих масс выглядит так:

Константа скорости реакции k показывает, с какой скоростью будут реагировать вещества, если их концентрации равны 1 моль/л, или их произведение равно 1. Константа скорости химической реакции зависит от температуры и не зависит от концентрации реагирующих веществ.
В законе действующих масс не учитываются концентрации твердых веществ, т.к. они реагируют, как правило, на поверхности, и количество реагирующих частиц на единицу поверхности при этом не меняется.
В большинстве случаев химическая реакция состоит из нескольких простых этапов, в таком случае уравнение химической реакции показывает лишь суммарное или итоговое уравнение происходящих процессов. При этом скорость химической реакции сложным образом зависит (или не зависит) от концентрации реагирующих веществ, полупродуктов или катализатора, поэтому точная форма кинетического уравнения определяется экспериментально, или на основании анализа предполагаемого механизма реакции. Как правило, скорость сложной химической реакции определяется скоростью его самого медленного этапа (лимитирующей стадии).
3. Давление
Концентрация газов напрямую зависит от давления . При повышении давления повышается концентрация газов. Математическое выражение этой зависимости (для идеального газа) — уравнение Менделеева-Клапейрона:
pV = νRT
Таким образом, если среди реагентов есть газообразное вещество, то при повышении давления скорость химической реакции увеличивается, при понижении давления — уменьшается .
Например. Как изменится скорость реакции сплавления извести с оксидом кремния:
при повышении давления?
Правильным ответом будет – никак, т.к. среди реагентов нет газов, а карбонат кальция – твердая соль, нерастворимая в воде, оксид кремния – твердое вещество. Газом будет продукт – углекислый газ. Но продукты не влияют на скорость прямой реакции.
4. Катализатор
Еще один способ увеличить скорость химической реакции – направить ее по другому пути, заменив прямое взаимодействие, например, веществ А и В серией последовательных реакций с третьим веществом К, которые требуют гораздо меньших затрат энергии (имеют более низкий активационный энергетический барьер) и протекают при данных условиях быстрее, чем прямая реакция. Это третье вещество называют катализатором .

Катализаторы – это химические вещества, участвующие в химической реакции, изменяющие ее скорость и направление, но не расходующиеся в ходе реакции (по окончании реакции не изменяющиеся ни по количеству, ни по составу). Примерный механизм работы катализатора для реакции вида А + В можно представить так:
A + K = AK
AK + B = AB + K
Процесс изменения скорости реакции при взаимодействии с катализатором называют катализом. Катализаторы широко применяют в промышленности, когда необходимо увеличить скорость реакции, либо направить ее по определенному пути.
По фазовому состоянию катализатора различают гомогенный и гетерогенный катализ.
Гомогенный катализ – это когда реагирующие вещества и катализатор находятся в одной фазе (газ, раствор). Типичные гомогенные катализаторы – кислоты и основания. органические амины и др.
Гетерогенный катализ – это когда реагирующие вещества и катализатор находятся в разных фазах. Как правило, гетерогенные катализаторы – твердые вещества. Т.к. взаимодействие в таких катализаторах идет только на поверхности вещества, важным требованием для катализаторов является большая площадь поверхности. Гетерогенные катализаторы отличает высокая пористость, которая увеличивает площадь поверхности катализатора. Так, суммарная площадь поверхности некоторых катализаторов иногда достигает 500 квадратных метров на 1 г катализатора. Большая площадь и пористость обеспечивают эффективное взаимодействие с реагентами. К гетерогенным катализаторам относятся металлы, цеолиты — кристаллические минералы группы алюмосиликатов (соединений кремния и алюминия), и другие.

Пример гетерогенного катализа – синтез аммиака:
В качестве катализатора используется пористое железо с примесями Al2O3 и K2O.
Сам катализатор не расходуется в ходе химической реакции, но на поверхности катализатора накапливаются другие вещества, связывающие активные центры катализатора и блокирующие его работу (каталитические яды). Их необходимо регулярно удалять, путем регенерации катализатора.
В биохимических реакция очень эффективными оказываются катализаторы – ферменты. Ферментативные катализаторы действуют эффективно и избирательно, с избирательностью 100%. К сожалению, ферменты очень чувствительны к повышению температуры, кислотности среды и другим факторам, поэтому есть ряд ограничений для реализации в промышленных масштабах процессов с ферментативным катализом.
Катализаторы не стоит путать с инициаторами процесса и ингибиторами.
Например , для инициирования радикальной реакции хлорирования метана необходимо облучение ультрафиолетом. Это не катализатор. Некоторые радикальные реакции инициируются пероксидными радикалами. Это также не катализаторы.
Ингибиторы – это вещества, которые замедляют химическую реакцию. Ингибиторы могут расходоваться и участвовать в химической реакции. При этом ингибиторы не являются катализаторами наоборот. Обратный катализ в принципе невозможен – реакция в любом случае будет пытаться идти по наиболее быстрому пути.
5. Площадь соприкосновения реагирующих веществ
Для гетерогенных реакций одним из способов увеличить число эффективных соударений является увеличение площади реакционной поверхности . Чем больше площадь поверхности контакта реагирующих фаз, тем больше скорость гетерогенной химической реакции. Порошковый цинк гораздо быстрее растворяется в кислоте, чем гранулированный цинк такой же массы.
В промышленности для увеличения площади контактирующей поверхности реагирующих веществ используют метод «кипящего слоя».
Например , при производстве серной кислоты методом «кипящего слоя» производят обжиг колчедана.
6. Природа реагирующих веществ
На скорость химических реакций при прочих равных условиях также оказывают влияние химические свойства, т.е. природа реагирующих веществ.
Менее активные вещества будут имеют более высокий активационный барьер, и вступают в реакции медленнее, чем более активные вещества.
Более активные вещества имеют более низкую энергию активации, и значительно легче и чаще вступают в химические реакции.
Более стабильные вещества — это, например, те вещества, которые окружают нас в быту, либо существуют в природе.
Например , хлорид натрия NaCl (поваренная соль), или воды H2O, или металлическое железо Fe.
Более активные вещества мы можем встретить в быту и природе сравнительно редко.
Например , оксид натрия Na2O или сам натрий Na в быту и в природе не не встречаем, т.к. они активно реагируют с водой.
При небольших значениях энергии активации (менее 40 кДж/моль) реакция проходит очень быстро и легко. Значительная часть столкновений между частицами заканчивается химическим превращением. Например, реакции ионного обмена происходят при обычных условиях очень быстро.
При высоких значениях энергии активации (более 120 кДж/моль) лишь незначительное число столкновений заканчивается химическим превращением. Скорость таких реакций пренебрежимо мала. Например, азот с кислородом практически не взаимодействует при нормальных условиях.
При средних значениях энергии активации (от 40 до 120 кДж/моль) скорость реакции будет средней. Такие реакции также идут при обычных условиях, но не очень быстро, так, что их можно наблюдать невооруженным глазом. К таким реакциям относятся взаимодействие натрия с водой, взаимодействие железа с соляной кислотой и др.
Вещества, стабильные при нормальных условиях, как правило, имеют высокие значения энергии активации.
http://zadachi-po-khimii.ru/obshaya-himiya/kolligativnye-svojstva-rastvorov.html
http://chemege.ru/kinetika/