ОСНОВЫ СТРОЕНИЯ И КИНЕТИКИ ФЕРМЕНТОВ В БИОЛОГИЧЕСКИХ СИСТЕМАХ — О. А. Науменко — 2017
2 Общие вопросы кинетики и термодинамики ферментативных реакций
2.1 Законы классической термодинамики в биохимии
В последние 30 лет наблюдается бурное развитие ферментативной кинетики. Ферментативная кинетика изучает изменение скорости ферментативных реакций в зависимости от концентрации фермента, концентрации субстрата, наличия активаторов, ингибиторов, изменения температуры и PH среды, а также механизмы регуляции активности фермента в ходе реакции.
2.1.1 Основные понятия кинетики и термодинамики. Термодинамические системы
Кинетика — это раздел физики, изучающий изменение параметров термодинамической системы в ходе различных термодинамических процессов.
Ферментативная кинетика изучает законы термодинамики: тепловой и энергетический обмены в биологических системах. При этом живые биологические системы, могут достигать динамического или стационарного состояния (динамического равновесия), в том случае, когда количество веществ и энергии, пришедшее в систему равно количеству вещества, ушедшего из системы.
Ферментативная кинетика изучает механизм, энергетику, регуляцию и изменение конформации ферментов в ходе ферментативных реакций. Изучением превращением энергии в биологических системах занимается раздел биохимии — биоэнергетика.
Химическая термодинамика использует законы термодинамики применительно к термодинамическим системам. Совокупность тел, участвующих в реакции называется термодинамической системой (ТС), а тела, не входящие в систему, называются внешним окружением. Термодинамические системы классифицируются на открытые и закрытые. Законы термодинамики применимы только к закрытым системам.
Закрытые системы обмениваются с окружающей средой только энергией, но не массой. Открытые системы, способны обмениваться с окружающей средой массой и энергией, а при определенных условиях достигать динамического равновесия или стационарного состояния, но не термодинамического равновесия с максимумом энтропии.
Биологические организмы — открытые термодинамические системы, которые обмениваются с окружающей средой энергией и веществом.
В термодинамически равновесных системах нельзя наблюдать макроскопических изменений. Их внутренняя энергия минимальна, и они находятся в состоянии полной беспорядочности. Живые системы достигают термодинамического равновесия только после смерти.
Все открытые системы вместе с их окружением образуют закрытую систему.
При наличии теплообмена и возможности хотя бы частичной диффузии между телами, составляющими систему, образуется термодинамическая система. Термодинамическая система может взаимодействовать со своим окружением, и это взаимодействие можно обнаружить по переносу тепла или совершению работы.
В том случае, когда взаимодействие системы со средой полностью отсутствует, система называется изолированной.
Если состояние термодинамической системы остается неизменным и причиной этого не является какой-либо внешний стационарный процесс, говорят, что система находится в равновесии. Если система состоит из одной фазы, то она гомогенная, в противном случае — гетерогенная.
Законы термодинамики применимы к закрытым системам. Поэтому для исследований и расчетов в биоэнергетике ввели понятие изолированной системы (минимальный обмен энергии).
Термодинамические системы можно классифицировать по совокупности их свойств. Свойства термодинамической системы — называются термодинамическими параметрами системы.
Экстенсивные свойства, такие как вес и объем, пропорциональны массе. Интенсивные свойства, такие как температура, давление, не зависят от массы. Состояние системы, находящейся в равновесии, можно описать совокупностью ее интенсивных свойств. Термодинамические параметры системы описывают только данное состояние, не учитывая предшествующих. Следовательно, изменение параметров при переходе системы из одного состояния в другое не зависит от пути реакции, а определяется только термодинамическими параметрами начального и конечного состояний.
Масса (М) и объем (V) — это общепринятые термины. Давление (Р) — характеризует взаимодействие с внешним окружением, измеряемое как сила, приходящаяся на единицу площади поверхности.
Температура (Т), которая определяется интенсивностью теплового движения молекул, образующих систему, не простое понятие, оно включает в себя понятие разности теплоты. Между телами различной температуры происходит теплоперенос, приводящий к выравниванию температур. Абсолютная шкала температур основана на втором законе термодинамики. Ее начало находится на абсолютном нуле (- 273,16о К).
При абсолютном нуле часть энергии любого вещества, которая зависит от температуры (тепловая энергия), равна нулю, хотя энергия частиц, составляющих вещество, при нулевой температуре, естественно, не исчезает.
2.1.2 Термодинамический процесс
Любое изменение параметров термодинамической системы, приводящее к изменению хотя бы одного термодинамического параметра, называется термодинамическим процессом (ТП).
Признаки равновесного термодинамического процесса. Если в ходе термодинамического процесса система проходит равновесные состояния, то при данных условиях работа, совершаемая самой системой, будет максимальна, а работа, свершаемая над системой, — минимальна, в таком случае говорят о равновесном ТП.
И наоборот, ТП, протекающий при некотором ограниченном воздействии на систему, определяется как неравновесный ТП. Работа, совершенная такой системой, меньше, чем максимальная работа в равновесном процессе.
Если единственным результатом обратного процесса в изолированной системе является возвращение системы из конечного состояния в исходное, то такой процесс называют обратимым ТП.
Если в результате прямой или обратной реакции в системе или в ее окружении имеют место длительные изменения термодинамических параметров системы, то ТП называют необратимым.
Причина необратимости в том, что процессы в ТС протекают через неравновесные состояния. Термодинамические параметры однозначны только для обратимых процессов, когда система находится в равновесии в любой момент времени и в каждой ее части. Если вывести систему из состояния устойчивого равновесия, то возникнет термодинамический процесс, препятствующий внешнему воздействию (принцип Ле Шателье — Брауна).
2.1.3 Первый закон термодинамики
Первый закон термодинамики гласит о том, что энергия в ТС не может быть получена из ничего и не может быть уничтожена, а может только превращаться из одного вида в другой. Это означает, что содержание энергии (U) в термодинамической системе увеличивается при совершении ТС работы (А) или передачи тепла (Q):
где ∆U — изменение энергии ТС; А — работа ТС; Q — теплота ТС.
В случае ТП, когда изменение энергии системы ∆U = 0,
Отсюда проистекает невозможность создания вечного двигателя первого рода.
Если над ТС не совершается никакой работы, т. е. ∆U = ∆Q, то при равновесном давлении (∆Р = 0) для объема V можно определить новую функцию:
где Н — энтальпия (или теплосодержание ТС); U — внутренняя энергия ТС; Р — давление в ТС; V — объем ТС.
Закон Гесса: теплота превращения в ТС не зависит от пути протекания ТП.
При экзотермическом процессе — выделенная ТС теплота считается отрицательной величиной (теплосодержание системы уменьшается), т. к. положительным условно считается тепло, полученное ТС (+Н)- это эндотермический процесс, сопровождающийся увеличением теплосодержания ТС. Протекающие в живых организмах анаболические процессы представляют собой эндотермические реакции, а катаболические — экзотермические.
2.1.4 Второй закон термодинамики
Все процессы в природе протекают в одном направлении, т. е. они необратимы. Необратимость в термодинамике означает, что ТП процесс не может идти в ТС в обратном направлении без изменения энтропии. Энтропия выражается следующим уравнением:
где ∆S — изменение энтальпии ТС; ∆Q — изменение теплоты ТС; Т — температура ТС.
Дифференциальное изменение энтропии равняется отношению элементарного количества теплоты к абсолютной температуре. Основываясь на втором законе термодинамике, все природные процессы, не противоречащие первому закону термодинамики, можно разделить на две группы:
— самопроизвольные ТП при данных условиях;
— не самопроизвольные ТП.
Невозможна реакция, дающая только перенос тепла от тела с более низкой температурой к телу с более высокой температурой. Это означает, что работа в ТС не может быть выполнена исключительно за счет тепловой энергии окружающей среды, другими словами, невозможно создание вечного двигателя второго рода.
При обратимых ТП в изолированной ТС энтропия остается неизменной, при необратимых процессах она возрастает. Если в результате необратимого процесса изолированная система приходит в равновесие, то ее энтропия достигает максимума. Следовательно, изменение энтропии определяет направление ТП и одновременно условия термодинамического равновесия.
Принцип постоянства или увеличения энтропии справедлив только для изолированной системы. В изолированной системе увеличение энтропии служит мерой необратимости процесса. Живые организмы сохраняют внутреннюю упорядоченность (постоянную энтропию), получая дополнительную энергию в виде пищевых веществ (химическая энергия) или солнечного света (электромагнитная энергия) из окружающей среды и возвращают в нее такое же количество энергии в другой форме, главным образом в форме тепла, которое рассеивается во всей
остальной Вселенной. Живые организмы непрерывно должны повышать свою энтропию для поддержания внутреннего порядка.
2.1.5 Характеристические функции состояния ТС
Характеристической функцией называется такая функция ТС, посредством которой могут быть выражены ее свойства. Под потенциалом понимают функцию, изменение которой связано с работой ТС. В термодинамике наиболее широко используют следующие характеристические функции:
1) изохорно-изотермический потенциал ТС — энергия Гельмгольца (∆F);
2) изобарно-изотермический потенциал ТС — энергия Гиббса (∆G);
3) внутренняя энергия ТС (∆U);
4) энтальпия ТС (∆Н);
5) энтропия ТС (∆S).
Внутренняя энергия (∆U) ТС определяется согласно уравнения:
где ∆U -изменение внутренней энергии ТС; Т — температура; ∆S — энтропия; Р — давление; ∆V — объем.
Энтальпия ∆Н — это изменение теплосодержания системы при постоянном давлении и энтропии.
где ∆Н — изменение теплосодержания ли энтальпии ТС; Т — температура; ∆S — энтропия; ∆Р — давление; V — объем.
Энтропия ∆S — это мера неупорядоченности ТС, величина, равная количеству теплоты, поступающей в ТС при постоянной температуре.
где ∆S -изменение энтропии ТС; Т — температура; ∆S — энтропия; ∆Q — изменение теплоты.
Энергия Гельмгольца — это свободная энергия ТС, идущая на совершение полезной работы при постоянном объеме и температуре (изохорно-изотермический потенциал).
где F — свободная энергия ТС; Т — температура; ∆S — энтропия; U — внутренняя энергия ТС.
Энергия Гиббса — это свободная энергия ТС, идущая на совершение системой полезной работы при постоянном давлении и температуре (изобарно- изотермический потенциал).
где G — энергия Гиббса; Т — температура; S — энтропия; Н — энтальпия.
Контрольные вопросы по изучаемой теме
1. Что изучает ферментативная кинетика?
2. Что такое термодинамический процесс и какие его виды?
3. Дайте понятие термодинамической системы?
4. Какие бывают термодинамические системы?
5. Что значит открытая термодинамическая система?
6. Сформулируйте первый закон термодинамики.
7. Сформулируйте второй закон термодинамики.
8. Термодинамический процесс и его виды.
9. Энтальпия и энтропия.
10. Характеристические функции в термодинамике.
2.2 Кинетика Михаэлиса — Ментена
2.2.1 Химическая кинетика. Понятие порядка реакции
Химическая кинетика — учение о химическом процессе, его механизме и закономерностях развитии во времени.
Химическая кинетика изучает скорость химической реакции с учетом различных условий: концентрации реагирующих веществ, температуры, pH среды, наличия или отсутствия катализаторов.
В химии одной молекуле вещества соответствует один моль вещества, а количество молей вещества обозначается коэффициентом реакции. В соответствии с законом действующих масс, порядок реакции определяется суммой степеней концентраций реагирующих веществ.
Например, в ходе химической реакции 1 моль вещества А превращается в 1 моль вещества В: 1 моль А —> 1 моль В
Это мономолекулярная реакция, или реакция первого порядка.
Если в ходе реакции, 1 моль вещества А взаимодействует с 1 молем вещества В и в результате их взаимодействия образуется 1 моль вещества С: А + В —> С, то эта реакция будет называться бимолекулярной или реакцией 2 порядка.
2.2.2 Кинетика Михаэлиса — Ментена
В 1835 году Ян Берцелиус впервые предположил, что реакции живого организма осуществляются благодаря «новой силе», которую он назвал «каталитической». Берцелиус сформулировал принцип катализа, включив в число каталитических агентов и фермент диастазу, которая катализировала гидролиз крахмал быстрее, чем серная кислота.
Предварительные эксперименты по изучению кинетики ферментативных реакций показали, что скорость ХФР, описываемой уравнением: Е + S —> Е + Р, не зависит от концентрации фермента и субстрата так, как в случае обычной химической реакции второго порядка.
Самая ранняя попытка математически описать ферментативные реакции была предпринята Дюкло в 1898 году. Браун (1902) и независимо от него Анри (1903) впервые выдвинули гипотезу об образовании в ходе реакции фермент — субстратного комплекса.
В 1913 году Михаэлис и Ментен опубликовали свою теорию общего механизма ферментативных реакций. Их уравнение стало фундаментальным принципом всех кинетических исследований ферментов.
Так, Михаэлис и Ментен предположили, что механизм обычной двухстадийной ХФР описывается схемой:
 (2.10),
(2.10),
где: Е — фермент; S — субстрат; ES — фермент — субстратный комплекс; Р — продукт ХФР; k1 — константа диссоциации первой стадии (реакция второго порядка);
к2 — константа диссоциации второй стадии распада фермент субстратного комплекса (реакция первого порядка).
В данной схеме k1 = кs, где кs — константа диссоциации фермент субстратного комплекса, а скорость ХФР определяется скоростью распада фермент — субстратного комплекса до продукта, т. е. скоростью второй стадии:
где: k2 — константа диссоциации второй стадии распада фермент субстратного комплекса (реакции первого порядка); v0 — начальная скорость ХФР; [ES] — концентрация фермент — субстратного комплекса.
Модель предполагает, что равновесие первой стадии ХФР между свободными ферментом, субстратом и фермент — субстратным комплексом устанавливается быстро по сравнению со скоростью всей ХФР (быстро устанавливающееся равновесие первой стадии к2 «к1, k-1). В этом случае вторая стадия реакции практически не влияет скорость первой стадии, и поэтому для выражения концентрации фермента можно воспользоваться константой диссоциации фермент — субстратного комплекса Ks.
 (2.12)
(2.12)
где Ks — константа диссоциации фермент субстратного комплекса; [Е] — концентрация фермента; [S] — концентрация субстрата; [ES] — концентрация фермент — субстратного комплекса.
Общая концентрация фермента в реакционной смеси выражается уравнением материального баланса по ферменту:
где [Е]0 — начальная концентрация фермента; Ks — константа диссоциации фермент субстратного комплекса; [Е] — концентрация фермента; [S] — концентрация субстрата;
[ES] — концентрация фермент — субстратного комплекса.
Тогда концентрация фермент — субстратного комплекса равна:
 (2.14)
(2.14)
Реакция достигает максимальной скорости, когда весь фермент находится в комплексе с субстратом:
где Vmax — максимальная скорость ХФР; ks — константа диссоциации фермент — субстратного комплекса; [Е]0 — начальная концентрация фермента; [S] — концентрация субстрата; [ES] — концентрация фермент — субстратного комплекса.
Это условие выполняется, если реакция протекает при избыточной концентрации субстрата: [S]0>> [Е]0. Из предыдущих уравнений получим:
 (2.16)
(2.16)
где v0 — начальная скорость ХФР; Vmax — максимальная скорость ХФР; Ks — константа диссоциации фермент — субстратного комплекса; [S] — концентрация субстрата.
Это и есть классическое уравнение Михаэлиса — Менте на, которым и сегодня пользуются для описания кинетики ХФР, а величина константы Михаэлиса при данных условиях Км = Ks и представляет собой меру сродства фермента субстрату. Максимальная скорость ХФР достигается тогда, года концентрация фермент — субстратного комплекса численно равна общей концентрации фермента (т. е. когда весь фермент связан с субстратом в комплекс).
В этом случае зависимость скорости ХФР от концентрации (Е) во многом зависит от соотношений концентрации фермента и субстрата. Если концентрация субстрата достигает максимальной и значительно превышает концентрацию фермента ([S]»[E]), то тогда скорость ХФР возрастает линейно прямо пропорционально концентрации фермента, т.е. чем выше концентрация [Е], тем выше будет скорость реакции.
2.2.3 Ограничения кинетики Михаэлиса-Ментена
Михаэлис и Ментен вывели уравнение с учетом двух условий: быстро устанавливающееся равновесие первой стадии и значительный избыток субстрата. Позднее было доказано, что уравнение справедливо, при выполнении семи условий или постулатов.
Постулаты выполнения уравнения Михаэлиса — Ментена:
1) в ходе реакции образуется кинетически устойчивый фермент-субстратный комплекс, который существует в определенной концентрации в течении определенного времени;
2) определяемая с помощью уравнения константа Ks является константой диссоциации фермент-субстратного комплекса: это справедливо, только если к2« к1, к-1;
3) концентрация субстрата не меняется в ходе реакции, то есть [S] = [S]0;
4) продукт реакции быстро отщепляется от фермента, то есть реакция двухстадийная;
5) вторая стадия реакции необратима. Так как это практически не выполнимо, мы принимаем во внимание только начальные скорости ХФР;
6) с каждым активным центром фермента связывается только одна молекула субстрата;
7) для всех реагирующих веществ вместо активностей можно использовать их концентрации.
2.2.4 Образование кинетически устойчивого фермент-субстратного комплекса (обоснование первого постулата)
К настоящему времени по данным рентгено — структурного анализа имеются сотни экспериментальных доказательств, демонстрирующих образование кинетически устойчивых фермент — субстратных комплексов в ходе ферментативных реакций. Хотя выдвигались также и другие теории, без образование в ХФР фермент — субстратного комплекса.
1. Теория телекинетических взаимодействий предполагает, что фермент увеличивает энергию молекулы субстрата с помощью каких-либо телекинетических взаимодействий (электростатическое притяжение или отталкивание, электромагнитное излучение и т. д.).
2. Теория упругих столкновений. Считается, что фермент передает энергию субстрату при упругих столкновениях. При этом молекула субстрата достигает энергии выше определенного уровня и может вступать в реакцию или разрушаться с образованием продуктов.
Несмотря на то, что для данных теорий были составлены кинетические уравнения, когда фермент отделен от субстрата растворителем (т. е. может происходить телекинез в передаче энергии субстрату), такие теории до сих пор не нашли экспериментального подтверждения.
Это означает, что образование кинетически устойчивого фермент- субстратного комплекса обязательно происходит во всех известных ферментативных реакциях. Поэтому первое условие, определяющее справедливость кинетики Михаэлиса — Ментена, можно считать доказанным.
2.3 Природа константы К в уравнении Михаэлиса — Ментена
2.3.1 Стационарная кинетика Бриггса и Холдейна (обоснование второго постулата)
Второй постулат формулирует, что константа Ks в уравнении (2.16) является константой диссоциации фермент-субстратного комплекса.
Бриггс и Холдейн в 1925 году доказали, что исходное уравнение Михаэлиса — Ментена справедливо только при условии к2 «к1, к-1 т. е. когда равновесие первой стадии (Е + S ⇄ES) устанавливается очень быстро по сравнению со скоростью установления равновесия второй стадии.
Поэтому такие кинетические механизмы, подчиняющиеся начальному условию Михаэлиса-Ментен и имеющие одну медленную стадию, относительно которой равновесия во всех других стадиях устанавливаются быстро, называются «быстрым равновесием».
Если к2 по порядку величины сравнима с к-1, то тогда используется кинетика Бриггса и Холдейна или кинетика стационарности: когда количество
образовавшегося фермент — субстратного комплекса равно количеству распавшегося. В этом случае уравнение начальной скорости ХФР будет следующим:
 (2.17)
(2.17)
Где V — скорость ХФР; Vmax — максимальная скорость ХФР; Км — константа Михаэлиса; [S] — концентрация субстрата; [Е] — концентрация фермента; К1 — константа диссоциации первой стадии (реакция второго порядка);
Данное уравнение аналогично уравнению (2.16), но оно расширяет область применимости исходного уравнения Михаэлиса-Ментена.
2.3.2 Природа константы К в уравнении Михаэлиса-Ментена
Если в ХФР выполняется условие быстро устанавливающегося равновесия первой стадии и k2«k1, к-1, то константа диссоциации К, которая будет найдена при исследовании зависимости скорости ХФР от концентрации субстрата, является константой диссоциации фермент — субстратного комплекса (первой стадии) Ks.
При выполнении условия стационарности: к2 = к1 (кинетика Бриггса и Холдейна), найденная тем же способом константа будет являться константой Михаэлиса, где Км= (к-1 + к2)/ к1.
В других реакциях, когда k1 «k2 константа Михаэлиса равняется k2/k1 и называется, согласно Ван Слайку, кинетической константой Кs.
При изменении условий реакции значение константы К также может изменяться. Так, например, в случае пероксидазы, при высокой концентрации субстрата — донора протонов, эта константа является кинетической константой (Кк). При уменьшении концентрации субстрата — донора протонов, константа превращается в константу Михаэлиса — Км, а при очень низких уровнях субстрата — донора протонов получаем константу диссоциации фермент — субстратного комплекса — Ks.
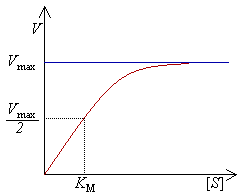
Поскольку значение константы Михаэлиса меняется в зависимости от условий реакции, константу Км также обозначают, как Км (каж.). График зависимости скорости ферментативной реакции от начальной концентрации субстрата S согласно уравнения (2.16) будет иметь гиперболическую форму (рисунок 2.1). На данном графике видно, что в начале реакции скорость реакции возрастает прямо пропорционально линейно с увеличением концентрации субстрата. Порядок реакции в этом случае будет первым. Скорость реакции на данном отрезке достигает максимальной (Vmax), когда весь субстрат будет связан с ферментом, и константа ХФР будет соответствовать Ks.
Рисунок 2.1 — График зависимости скорости ферментативной реакции от концентрации субстрата S.

Далее при нарастании концентрации субстрата, график меняется с прямой на параболу. В этом случае порядок реакции становится дробным (п=1/2). В этой части реакции Sрасходуется, концентрация его уменьшается и становится [S] = Км, а скорость реакции равняется 1/2 от Vmax. При дальнейшем увеличении концентрации субстрата S, скорость реакции не увеличивается, а порядок реакции становится нулевым (n = 0), а скорость реакции V= const.
Таким образом, физический смысл константы Михаэлиса (Км) состоит в том, что это такая концентрация субстрата [S] при которой, скорость ХФР составляет 1/2 от максимальной.
Значение констант для каждой ферментативной реакции определяется экспериментально и их значение зависит от pH — среды, температуры, присутствия ингибитора (активатора). Если для фермента определена Vmах и Км, то можно рассчитать оптимальную концентрацию субстрата S.
2.3.3 Методы графического определения кинетических параметров
Для решения практических задач энзимологии график гиперболический зависимости скорости ферментативной реакции от концентрации субстрата S является весьма неудобным. Поэтому на практике используются методы линеаризации экспериментальных данных.
Наиболее часто для определения кинетических параметров ХФР используются координаты Лайнуивера — Берка (1/V и 1/S) и координаты Диксона — Ида (1/V и [S]).
Метод Лайнуивера — Берка называется методом двойных обратных величин. График зависимости скорости ХФР от концентрации субстрата имеет линейный вид и представлен на рисунке 2.2.
Рисунок 2.2 — График зависимости скорости ферментативной реакции от концентрации субстрата S по методу Лайнуивера — Берка.

Определение обратных значений (1/V) скорости ХФР в координатах Лайнуивера — Берка ведется по формуле 2.18:
 (2.18)
(2.18)
где V — скорость ХФР; Vmах — максимальная скорость ХФР; Км — константа Михаэлиса; [S] — концентрация субстрата.
Контрольные вопросы по изучаемой теме
1. Порядок реакции и методы его определения.
2. История развития ферментативной кинетики.
3. Уравнение Михаэлиса-Ментена.
4. Ограничение кинетики Михаэлиса-Ментена.
5. Образование кинетически устойчивого фермент-субстратного комплекса.
6. Природа константы К в уравнения Михаэлиса-Ментена.
7. В чем физический смысл константы Михаэлиса.
8. Какой формы график зависимости скорости ХФР от концентрации субстрата согласно уравнения Михаэлиса-Ментена?
9. Какие координаты в зависимости Лайнуивера — Берка?
10. Что такое кинетическая константа?
11. Какие имеются доказательства первого постулата?
2.4 Кинетический анализ двухстадийных ферментативных реакций, не подчиняющихся уравнению Михаэлиса-Ментена
Третий постулат ферментативной кинетики гласит: концентрация субстрата не меняется в ходе реакции, то есть [S]t = [S]0. Это означает наличие достаточно большого избытка субстрата. Однако во многих случаях это условие не выполняется. С одной стороны, большой избыток субстрата не используется в реакциях «in vitro» с некоторыми ферментами из-за частого ингибирования субстратом ферментативной активности самого фермента. Обычно не достигается избыток субстрата и «in vivo».
В ферментативных реакциях, где субстрат не находится в избытке и, следовательно, его концентрация меняется с течением времени в ходе реакции, константа диссоциации примет вид:
 (2.19)
(2.19)
где Ks — константа диссоциации фермент — субстратного комплекса; [S]0 — начальная концентрация субстрата; [ES] — концентрация фермент — субстратного комплекса;
[Е]0 — концентрация фермента в начале реакции; [Р] — концентрация продукта ХФР.
Начальная скорость реакции тогда будет:
 (2.20)
(2.20)
где Vmax — максимальная скорость ХФР; Ks — константа диссоциации фермент — субстратного комплекса; [S]t -концентрация субстрата на данный момент времени.
2.4.1 Изменение концентрации субстрата в ходе ферментативной реакции (обоснование третьего постулата)
Уравнение (2.20) можно решить, используя два разных варианта условий ХФР, когда [S]0 ≠ [S]t:
1) если это неравенство выполняется из-за больших значений t, т. е. когда более 5% от начальной концентрации субстрата израсходовано за время реакции (в начале реакции условие избытка субстрата выполнялось);
2) если концентрацией фермента нельзя пренебречь по сравнению с концентрацией субстрата и, таким образом, нужно принимать во внимание концентрацию фермент — субстратного комплекса (время реакции мало, но условие избытка субстрата не выполнено изначально).
2.4.1.1 Решение первого варианта: Если t велико, a [ES] «[S]0, уравнение (2.20) переходит в следующее:
 (2.21)
(2.21)
где Ks — константа диссоциации фермент — субстратного комплекса; [S]0— начальная концентрация субстрата; [ES] — концентрация фермент — субстратного комплекса;
[Е]0 — концентрация фермента в начале реакции; [Р] — концентрация продукта ХФР.
Для значения концентрации субстрата [S]t, которая меняется в ходе реакции, удовлетворительным приближением служит значение ([S]0+[S]t)/2. Тогда среднюю скорость можно выразить как:
 (2.22)
(2.22)
где Vmax — максимальная скорость ХФР; Км — константа Михаэлиса; [S]0— начальная концентрация субстрата; [S]t -концентрация субстрата на данный момент времени.
2.4.1.2 Решение второго варианта. Когда условие избытка субстрата не выполнено изначально, но расход субстрата не превышает 5%. В этом случае можно пренебречь концентрацией продукта [Р], но нельзя не учитывать концентрацию фермент-субстратного комплекса [ES],
 (2.23)
(2.23)
где Ks — константа диссоциации фермент — субстратного комплекса; [ES] — концентрация фермент — субстратного комплекса; [Е] — концентрация фермента;
Квадратное уравнение относительно концентрации субстрата:
 (2.24)
(2.24)
где Ks — константа диссоциации фермент — субстратного комплекса; [S]0 — концентрация субстрата в начале реакции; [S] — концентрация субстрата;
а — коэффициент, который равен:
где Ks — константа диссоциации фермент — субстратного комплекса; [S]0 — концентрация субстрата в начале реакции; [S] — концентрация субстрата.
Решением данного квадратного уравнения является
 (2.26)
(2.26)
где Ks — константа диссоциации фермент — субстратного комплекса; [S]0 — концентрация субстрата в начале реакции; [S] — концентрация субстрата; а — коэффициент.
 (2.27)
(2.27)
Тогда скорость реакции V можно выразить уравнением
 (2.28)
(2.28)
где к2 — константа диссоциации второй стадии; [S]0— начальная концентрация субстрата; [ES] — концентрация фермент — субстратного комплекса; [Е]0 — концентрация фермента в начале реакции; Vmax — максимальная скорость ХФР.
Выражая из последнего уравнения λ, получим
 (2.29)
(2.29)
где V — скорость ХФР; Vmax — максимальная скорость ХФР.
При сравнении значений, рассчитанных данным методом, со значениями, полученными из точного, проинтегрированного уравнения Михаэлиса — Ментена, оказывается, что ошибка в определении Км составляет только 1% и 4% при расходовании 30% и 50% субстрата соответственно. Следовательно, данными уравнениями можно пользоваться для расчета кинетических параметром ХФР при больших промежутках времени реакции и расходовании субстрата.
2.4.2 Методика нахождения кинетических констант при условии [S]0 ≠ [S]t
Методика нахождения констант в данном случае выглядит следующим образом.
1. В предварительных экспериментах (при избытке субстрата) определяют Vmах;
[S]0 определяют скорости реакции при разных концентрациях субстрата;
3. По уравнению (2.29) рассчитываем параметр λ для каждой вычисленной скорости;
4. По вычисленным параметрам λ определяем Ks.
По точке пересечения данной прямой с осью абсцисс находим Ks, тангенс угла наклона равен 1/[Е]0. Таким образом, изучение зависимости скорости ферментативной реакции от концентрации субстрата в условиях [E]0
[S]0позволяет определить абсолютную концентрацию активных центров фермента.
Контрольные вопросы по изучаемой теме
1. Что гласит третий постулат ферментативной кинетики?
2. Приближенное решение уравнения Михаэлиса-Ментен в случае больших времен протекания реакции.
3. Какие условия для первого варианта приближенного решения уравнения Михаэлиса-Ментен?
4. Какие условия для второго варианта приближенного решения уравнения Михаэлиса-Ментен?
5. Какова методика нахождения кинетических констант при условии [S]0 ≠ [S]t?
2.5 Влияние обратимых эффекторов на кинетику ферментативной реакции
2.5.1 Механизмы влияния обратимых эффекторов на кинетику ферментативной реакции
Вещества, изменяющие каталитическую активность фермента, называются эффекторами.
Взаимодействие фермента с эффектором представляет собой химическую реакцию и поэтому может быть полностью обратимым, частично обратимым или практически необратимым. Если процесс ингибирования необратим, то кинетическая реакция не подчиняется механизму Михаэлиса — Ментен, основой которого является наличие равновесия между свободной и связанной формами фермента.
В случае обратимых ингибиторов для описания кинетики ферментативной реакции можно воспользоваться уравнением Михаэлиса-Ментена. По влиянию ингибиторов на параметры ХФР их классифицируют:
а) конкурентные — ингибиторы, в присутствии которых повышается Км, a Vmax не меняется. Вызываемый такими ингибиторами эффект можно частично или полностью снять путем повышения концентрации субстрата. Примером может служить обратимое конкурентное ингибирование фермента сукцинилдегидрогеназы малоновой кислотой, субстратом которой является янтарная кислота (рисунок 2.3);
Рисунок 2.3 — Обратимое конкурентное ингибирование фермента сукцинилдегидрогеназы малоновой кислотой

б) неконкурентные — ингибиторы, инактивирующие фермент или фермент — субстратный комплекс путем уменьшения Vmax, но не влияющие на Км. В этом случае повышение концентрации субстрата не приводит к повышению скорости реакции. К неконкурентным ингибиторам относятся ионы тяжелых металлов, обратимо реагирующие с -SHгруппами цистеинов.
Многие из конкурентных ингибиторов по своей химической природе близки субстратам. Такие ингибиторы называются субстратными аналогами.
Неконкурентные ингибиторы связываются с аллостерическим центром фермента.
2.5.2 Кинетика ферментативных реакций, протекающих с участием эффектора
В общем случае влияние обратимого эффектора на двухстадийную ферментативную реакцию может быть передано схемой:
 (2.30)
(2.30)
где Е — фермент; S — субстрат; Р — продукт реакции; ks — константа диссоциации фермент — субстратного комплекса; к2 — константа диссоциации второй стадии;
кэ — константа диссоциации комплекса фермент — эффектор; ЭЕ — комплекс фермент — эффектор; ЭES — тройной комплекс эффектор — фермент — субстрат.
Для начальной скорости ферментативной реакции (при условии [S]0, [Э]0>> [Е]0, где [Э] — концентрация эффектора, а — коэффициент, показывающий во сколько разменяется скорость первой стадии в присутствии эффектора, а β — коэффициент, изменения скорости второй стадии при установившихся равновесиях первой и второй стадиях ХФР, справедливо:
 (2.31)
(2.31)
где [S]0 — начальная концентрация субстрата; [ES] — концентрация фермент — субстратного комплекса; [Е]0 — концентрация фермента в начале реакции; [Э] — концентрация эффектора; Ks — константа диссоциации фермент — субстратного комплекса; к2 — константа диссоциации второй стадии; кэ — константа диссоциации комплекса фермент — эффектор; а — коэффициент изменения скорости первой стадии; β — коэффициент, изменения скорости второй стадии.
В зависимости от численных значений α и β эффектор может выступать в роли либо ингибитора, либо активатора ферментативной реакции. Выделяют следующие виды ингибирования:
— полное конкурентное ингибирование;
— полное неконкурентное ингибирование;
2.5.2.1 Полное конкурентное ингибирование (α —> ∞, β не имеет определенного смысла), скорость ХФР определяется уравнением:
 (2.32)
(2.32)
где [S]0 — начальная концентрация субстрата; [Е]0 — концентрация фермента в начале реакции; [I] — концентрация ингибитора; кs — константа диссоциации фермент — субстратного комплекса; к2 — константа диссоциации второй стадии; ki — константа диссоциации комплекса фермент — ингибитор.
Зависимость в координатах Лайнуивера — Берка имеет вид пучка прямых, пересекающихся на оси ординат (рисунок 2.4).
Рисунок 2.4 — График зависимости при полном конкурентном ингибировании

Если отложить экспериментальные данные в координатах (КМ(Каж), [I]), константу конкурентного ингибирования Кi можно определить по формуле 2.33:
 (2.33)
(2.33)
где Км — константа Михаэлиса; [I] — концентрация ингибитора; кs — константа диссоциации фермент — субстратного комплекса; ki — константа диссоциации комплекса фермент — ингибитор.
2.5.2.2 Полное неконкурентное ингибирование (α —> 1, β =0). В этом случае, скорость ХФР определяется уравнением:
 (2.34)
(2.34)
где [S]0 — начальная концентрация субстрата; [Е]0 — концентрация фермента в начале реакции; [I] — концентрация ингибитора; кs — константа диссоциации фермент — субстратного комплекса; к2 — константа диссоциации второй стадии; ki — константа диссоциации комплекса фермент — ингибитор.
Зависимость в координатах Лайнуивера — Берка имеет вид пучка прямых, пересекающихся на оси абсцисс (рисунок 2.5).
Рисунок 2.5 — График зависимости при полном неконкурентном ингибировании

Если представить экспериментальные данные в координатах (1/kkat, [I], константу неконкурентного ингибирования К, можно определить, зная величину кк:
 (2.35)
(2.35)
где кк — каталитическая константа; [I] — концентрация ингибитора; к2 — константа диссоциации второй стадии; кi — константа диссоциации комплекса фермент — ингибитор.
2.5.2.3 Бесконкурентное ингибирование (α = β ≤ 1). В этом случае скорость ХФР:
 (2.36)
(2.36)
где [S]0 — начальная концентрация субстрата; [Е]0 — концентрация фермента в начале реакции; [I] — концентрация ингибитора; кs — константа диссоциации фермент — субстратного комплекса; к2 — константа диссоциации второй стадии; кi — константа ингибирования; α — коэффициент изменения скорости первой стадии; β — коэффициент, изменения скорости второй стадии.
Значения констант kkat и КМ(каж) ферментативной реакции при увеличении концентрации эффектора уменьшаются в одинаковой степени, поэтому графики в
координатах Лайнуивера — Берка имеют вид семейства параллельных прямых (рисунок 2.6).
Рисунок 2.6 — График зависимости при бесконкурентном ингибировании Для анализа уравнения выражение для каталитической константы

 (2.37)
(2.37)
где кк — каталитическая константа; [I] — концентрация ингибитора; к2 — константа диссоциации второй стадии; кi — константа ингибирования; α — коэффициент изменения скорости первой стадии, удобно преобразовать к следующему виду:
 (2.38)
(2.38)
где кк — каталитическая константа; [I] — концентрация ингибитора; к2 — константа диссоциации второй стадии; кi — константа ингибирования; α — коэффициент изменения скорости первой стадии.
Из графика, построенного в координатах (1/(kkat/k2 -1), 1/[I]), можно раздельно найти значения α и Кi).
2.5.2.4 Неконкурентная активация (α = 1, β >1). В этом случае субстрат и активатор связываются независимо с активны центром, образуя тройной комплекс (фермент — субстрат — активатор), что приводит к увеличению скорости образования продукта ХФР. Поэтому начальная скорость ХФР определяется выражением:
 (2.39)
(2.39)
где [S]0 — начальная концентрация субстрата; [Е]0 — концентрация фермента в начале реакции; [А] — концентрация активатора; кs — константа диссоциации фермент — субстратного комплекса; к2 — константа диссоциации второй стадии; кА — константа активирования; β — коэффициент, изменения скорости второй стадии.
Зависимость в координатах Лайнуивера — Берка имеет вид пучка прямых, пересекающихся на оси абсцисс.
2.5.2.5 Смешанные типы ингибирования и активации (α ≠ 1, β ≠ 1). В случае смешанных типов ингибирования или активации графики в координатах Лайнуивера — Берка имеют вид пучка прямых, соответствующих различным концентрациям эффектора, и пересекающихся в общей точке в правом верхнем, левом верхнем или левом нижнем квадранте в зависимости от значений α и β (рисунок 2.7).
Рисунок 2.7 — Графики зависимости при смешенном типе ингибирования

Координаты точки пересечения пучка прямых определяется значениями констант α и β.
Контрольные вопросы по изучаемой теме
1. Какие могут быть варианты взаимодействия фермента с эффектором?
2. Что обозначает обратимое ингибирование?
3. Что обозначает не обратимое ингибирование?
4. Что обозначает конкурентная активация?
5. Что обозначают коэффициенты α и β?
6. Как будет выглядеть график зависимости при смешенном типе ингибирования?
7. Как будет выглядеть график зависимости при неконкурентной активации?
8. Как будет выглядеть график зависимости при бесконкурентном ингибировании?
9. Как будет выглядеть график зависимости при полном неконкурентном ингибировании?
10. Как будет выглядеть график зависимости полном конкурентном ингибировании?
2.6 Ингибирование и активация избытком субстрата
2.6.1 Механизмы ингибирования субстратом
Для многих ферментов установлено, что при добавлении избытка субстрата скорость катализируемой реакции далее не увеличивается и не остается постоянной при концентрации выше насыщающей, а понижается, т. е. развивается ингибирование. Это явление было названо ингибированием избытком субстрата.
Что касается его механизмов, можно предложить пять различных типов ингибирования:
1) субстрат связывается с ферментом двумя или более группами, и только субстрат, связанный таким образом, может принимать участие в реакции. Если присутствует много молекул субстрата, то может возникнуть такая ситуация, что на одном связывающем центре фермента связывается одна молекула субстрата, в то время как на другом участке связывается другая молекула субстрата вместо того, чтобы взаимодействовать с другой группой первой молекулы субстрата. Так как в реакции может принимать участие только молекула, связанная с ферментом двумя точками, молекула фермента, удерживающая две молекулы субстрата, будет неактивна, т. е. будет развиваться ингибирование;
2) при высокой концентрации субстрат может связываться с другим участком, отличающимся от активного центра белка. В этом случае он может ’’неконкурентно’’ ингибировать скорость распада субстрата, связанного в активном центре;
3) фермент требует для проявления активности присутствия необходимого активатора. Если субстрат способен образовывать комплекс с активатором, избыток субстрата удаляет активатор, таким образом уменьшая активность фермента;
4) для реакции с двумя или несколькими субстратами избыток одного из субстратов способен связываться с участком связывания другого, ингибируя реакцию;
5) увеличение концентрации субстрата выше определенного предела неспецифически ингибирует реакцию вследствие увеличения ионной силы.
2.6.2 Кинетический анализ ХФР при ингибировании субстратом
Для многих ферментов зависимость v от [S]0можно количественно описать, исходя из предположения об образовании тройного комплекса ES2, не обладающего ферментативной активностью:
 (2.39)
(2.39)
где Е — фермент; S — субстрат; Р — продукт реакции; ES — фермент — субстратный комплекс; ES2 — тройной фермент — субстратный комплекс; Ks‘— константа диссоциации тройного фермент — субстратного комплекса; к2 — константа диссоциации второй стадии.
В этом случае уравнение скорости ферментативной реакции будет иметь вид:
 (2.40)
(2.40)
где [S]0 — начальная концентрация субстрата; [Е]0 — концентрация фермента в начале реакции; Ks — константа диссоциации фермент — субстратного комплекса;
к2 — константа диссоциации второй стадии.
Анализ уравнения разбивают на две части:
1) при низких концентрациях субстрата ([S]02«Ks‘) уравнение (2.40) упрощается до классического уравнения Михаэлиса — Ментена (не наблюдается ингибирование субстратом). Построением графика в координатах Лайнуивера — Берка находим к2 и Ks;
2) при высоких концентрациях субстрата ([S]0» Ks) уравнение (2.40) примет вид:
 (2.41)
(2.41)
где v — скорость ХФР; [S]0— начальная концентрация субстрата; [Е]0 — концентрация фермента в начале реакции; Ks — константа диссоциации фермент — субстратного комплекса; Ks‘— константа диссоциации тройного фермент — субстратного комплекса; к2 — константа диссоциации второй стадии.
Уравнение (2.41) можно привести к виду:
 (2.42)
(2.42)
и построив зависимость 1/v от [S]0, найти значения к2 и Ks‘. Если в обеих областях найденные значения к2 совпадают, значит схема (2.39) применима для формального описания кинетики изучаемой ферментативной реакции.
Во второй модели тройной комплекс ES2 обладает активностью, но меньшей по сравнению с фермент-субстратным комплексом ES (β n KsKs‘, 1/v). Значение n, при котором график линеен, соответствует зависимости (2.44), и, следовательно, модели (2.45) с найденным значением n.
Мы рассмотрели 3 схемы возможного ингибирования ферментативной активности избытком субстрата. Чтобы выбрать механизм ингибирования, сначала надо построить зависимость экспериментально найденных значений скорости в координатах (v, lg[S]0).
Симметричный вид полученной колоколообразной кривой указывает на отсутствие ферментативной активности комплекса SES (рисунок 2.8).
Рисунок 2.8 — Зависимость в координатах для всех моделей ингибирования субстратом

Если же правая ветвь графика (при больших концентрациях субстрата) более пологая по сравнению с левой ветвью, это говорит о ферментативной активности
тройного комплекса (0 1. Анализ экспериментальных данных проводится аналогично (2.43). Единственное отличие заключается в том, что удобнее строить график в координатах (1/(кэфф/к2-1), 1/[S]0) согласно зависимости:
 (2.47)
(2.47)
где [S]0 — начальная концентрация субстрата; Ks — константа диссоциации фермент — субстратного комплекса; Ks‘— константа диссоциации тройного фермент —субстратного комплекса; β — коэффициент, изменения скорости второй стадии; к2 — константа диссоциации второй стадии.
Контрольные вопросы по изучаемой теме
1. Какие существуют варианты взаимодействия между ферментом и избытком субстрата?
2. Напишите схему реакции при ингибирование избытком субстрата с образованием тройного комплекса субстрат — фермент -субстрат?
3. Как найти скорость ХФР в этом случае?
4. Напишите схему реакции при активации субстратом.
5. Как определить кинетические параметры ХФР в этом случае?
2.7 Факторы, влияющие на ферментативную активность. Влияние pH и температуры на кинетику ферментативных реакций
2.7.1 Факторы, влияющие на ферментативную активность
На каталитическую активность ферментов влияют многие факторы, которые могут изменять строение или химическую природу ферментов. К числу таких факторов относятся:
3) силы, действующие в текучих средах (гидродинамические силы, гидростатическое давление и поверхностное натяжение);
4) химические агенты (спирт, мочевина или пероксид водорода);
5) облучение (свет, звук, ионизирующая радиация).
Иногда снижение каталитической активности, вызванное, например, изменением pH, обратимо. В таких случаях возврат к первоначальным условиям сопровождается восстановлением активности фермента. В известном смысле такая ситуация аналогична рассмотренному случаю обратимого ингибирования. Небольшие изменения одного из перечисленных выше факторов, только слегка сдвигают равновесие (или квазистационарное состояние), характерное для данной ферментативной реакции. В общем случае отклонение от условий, типичных для биологического окружения нативного фермента, должно быть относительно небольшим (или кратковременным). В противном случае возрастает вероятность инактивации фермента.
2.7.2 Обратимое влияние температуры на каталитическую активность фермента
Граница между «обратимой» и «необратимой» инактивацией белков не всегда четко определена. Например, подвергнутый кратковременному нагреванию фермент при охлаждении до свойственной ему «рабочей» температуры может полностью восстановить свою активность. С другой стороны, более продолжительное нагревание при той же температуре или столь же кратковременная термообработка при более высокой температуре могут привести к тому, что при последующем охлаждении активность фермента восстановится лишь частично. Такое поведение белков — ферментов становится понятным, если учесть связь между их строением и функцией, влияние молекулярной динамики на функцию белка и возможность разрыва некоторых слабых связей в нативной структуре фермента при изменении условий среды.
При высокой температуре, когда начинает доминировать процесс термической инактивации фермента, нарушаются зависимости скорости реакции от температуры.
Так ферментативные реакции имеют колоколообразную зависимость скорости реакции от температуры, что объясняется наложением двух эффектов — возрастанием скорости реакции при увеличении температуры и ускорением тепловой денатурации белковой молекулы, приводящей к инактивации фермента при высоких температурах. Денатурация большинства белков начинается в диапазоне температур от 45° С до 50° С и завершается очень быстро при 55° С.
2.7.3 Влияние pH на кинетику ферментативных реакций в растворах
Ферменты, как и все белки, состоят из аминокислот. В зависимости от pH радикалы некоторых аминокислот, а значит, и белок в целом могут приобретать заряд. Заряженные группы часто входят в состав активных центров ферментов, так как в основе целого ряда механизмов ферментативного катализа лежит катализ
кислотного или основного типа. Необходимым условием для осуществления кислотного или основного катализа может быть наличие определенного заряда на ионизируемых группах активного центра. Отсюда следует, что каталитически активная форма фермента существует только в одном строго определенном состоянии ионизации, и в зависимости от pH в нее может превращаться большая или меньшая часть всего имеющегося в смеси фермента (рисунок 2.9).
Рисунок 2.9 — Зависимость активности ферментов от pH

При значениях pH, значительно отличающихся от оптимальных, происходит нарушение сил, стабилизирующих конформацию нативного белка — фермента, что может привести к его денатурации — утрате третичной конформации, и соответственно потери каталитической активности. В этом случае даже после восстановления оптимального pH ренатурация фермента становится маловероятной.
Контрольные вопросы о изучаемой теме
1. Какие факторы влияют на ферментативную активность?
2. Влияние pH на кинетику ферментативных реакций в растворах.
3. Как изменяется скорость ХФР при изменении температуры?
4. Какие процессы происходят при денатурации в структуре фермента?
Это и есть уравнение Михаэлиса-Ментен.
Вариант 3.
Уравнение Михаэлиса – Ментена, его вывод и анализ приложенности к описанию зависимости начальной скорости ферментативных реакций от концентрации субстрата.
ВЫВОД И АНАЛИЗ УР МИХАЭЛИСА-МЕНТЕН
Михаэлис и Ментен предположили, что мех-м ферм р-й описывается моделью:
При формулировке кинетического выражения для скорости ферментативной реакции Михаэлис и Ментен сделали три допущения:
1) Стационарное состояние реакции в момент равновесия, когда скорости образования и расходования ES равны;
2) Весь фермент в условиях насыщающих концентраций субстрата превращается в энзимсубстратный комплекс ES;
3) Если весь фермент в виде ES, то скорость реакции максимальна и Vmax=k2[ES].
Образование ES: [ES]=k1[S][E] (I)
Расходование ES: [ES]=k-1[ES]+k2[ES] (II)
Приравнивая выражения (I) и (II) и сокращая обе части на k1 получаем:
[S][E] = [ES](k-1 + k2)/k1 = [ES]Km, где Km = (k-1 + k2)/k1
Выразим равновесную концентрацию [E] через начальную [Eo]: [E] = [Eo] — [ES]
[S]([Eo]-[ES])= [ES]Km, переносим [S] в правую часть выражения и делим обе части на [ES]:
Поскольку трудно (если не невозможно) измерить [ES], произведем замену с учетом того, что в насыщающих концентрациях [S] весь [Eo] перейдет в [ES] и максимальная скорость при этом будет равна Vmax=k2[ES]=k2[Eo].
В это же время скорость реакции равна V=k2[ES]. Через отношение этих скоростей выразим [Eo]/[ES]:
В уравнении (III) произведем замену отношения [Eo]/[ES] на Vmax/V и получаем:
Это и есть уравнение Михаэлиса-Ментен.
Ограничения кинетики Михаэлиса-Ментен
Михаэлис и Ментен вывели уравнение с учетом двух предположений (быстро устанавливающееся равновесие и избыток субстрата). Позднее было показано, что уравнение справедливо, то есть хорошо описывает реакцию, при выполнении всех следующих условий.
7 основных постулатов для выполнения уравнения Михаэлиса-Ментен.
1. В ходе реакции образуется кинетически устойчивый фермент-субстратный комплекс.
2. Определяемая с помощью уравнения константа Кs является константой диссоциации фермент-субстратного комплекса: это справедливо, только если k2
V = Vmax[S]/(Km+[S]) — ур михаэлиса-ментен
Граф зав-сть для ур имеет вид: Кривая уравнения Михаэли-са-Ментен: гиперболическая зависимость начальных скоростей катализируемой ферментом реакции от концентрации субстрата.
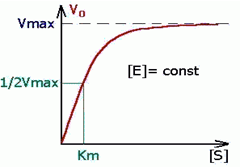
Константа Михаэлиса измеряется в молях на литр и бывает от 10-2 до 10-7, чем меньше Кm, тем активнее фермент. При V=1/2Vmax, имеем Km = [S]. Однако, определение Vmax затруднительно по асимптоте. Для устранения этого неудобства ЛАЙНУИВЕР и БЭРК приравняли обратные зависимости левой и правой частей уравнения.
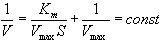
Своеобразие проявления второго закона термодинамики в биологических системах.
— ограничивает переход какой –либо Е в работу или в другой тип энергии. Ни какой тип Е не может перейти в работу с КПД – 100%.
1) не возможно перевести тепло от более холодной системы к более горячей без соответствующих изменений в этих системах и в окр. среде, т.е нельзя закипятить стакан с водой в холодильнике.
2) Самопроизвольно могут протекать лишь те процессы, которые связаны с переносом Е от более высокого уровня к более низкому, т.е. по градиенту.
3) Невозможно совершить работу против градиента без соответствующих изменений.
Градиент — векторная величина, разность величин того или иного параметра в 2-х точках, отнесенных к расстоянию между ними. Градиент концентрации – это концентрация «в» и «извне» на толщину мембраны (осмотич. градиент, концентрационный гр. (транспорт ионов и др. в-в), электрический гр.).
Все процессы, протекающее в природе, подчиняются первому закону ТД, однако не всякий возможный процесс осуществим на практике. Исходя из первого закона, нельзя определить направление самопроизвольного процесса. Второй закон ТД позволяет предсказать направление пр-са при заданных усл.. В отл. от 1 закона ТД он не носит всеобщего характера и применим лишь к сист., сост. из большого числа частиц.
14. Доказательства применимости второго закона ТД к биосистемам.
Применимость второго закона ТД к биосистемам:
1. Второй закон ТД был сформулирован для характристики изолированных систем. Реальные био системы являются открытыми.
2. Значение энтропии строго определено для равновесного состояния. Био системы в своем развитии проходят через целый ряд неравновесных состояний.
Развитие орг-мов сопровождаются усложнением их организации – это самопроизвольное ↓ энтропии живых систем. В реальных усл. развитие орг-мов, сопр-ся ↓ общей величины их энтропии за счет того, что в др. участках внешней среды идут сопряженные процессы с обр-нием положительной энтропии. Суммарное изменение энтропии в системе организм + внешняя среда всегда положительно.
Био системы характеризуются наличием большого кол-ва градиентов (осмотический, электрический, концентрационный). Градиент какого-либо т/д параметра изменяется с расстоянием. Биосистема способна совершать работу, если в ней имеется градиент. Градиент – своеобразное депо энергии.
Совершение работы в системе связано с реализацией этой свободной энергии. Если совершается работа, то градиент, за счет Е которого это происходит, ↓, но параллельно возникает другой градиент противоположной направленности. При необратимых пр-сах величина второго градиента будет меньше, чем величина первого.
Вариант 1.
Закон термодинамики, закон Гисса, термодинамическое равновесие, стационарные системы.
8. Первый закон ТД в биологии; доказательства его применимости к живым системам. Своеобразие проявления первого закона ТД в биосистемах.
Закон: работа совершаемая системой = разности м/у количеством теплоты, сообщаемой системой и изменением её внутр. Е: ∆А = ∆Q — ∆U. Закон – это количественная форма закона сохранения энергии. Кол-во теплоты, поступающей в систему расходуется на ↑ внутр. Е системы за вычетом совершенной работы.
Внутренняя Е (U) – сумма (совокупность) всех типов Е и взаимодействий входящих в систему частиц. (Е вращательного движения атомов, Е взаим-вия водородных атомов).
Работа биоситемы может совершаться за счёт энтропии и внутр. Е (но не внеш. теплоты, т.к. если бы можно было за счёт притока из вне биосист. нагревались бы 1740С, см. ниже в этом же вопросе – это типа своеобразие закона) (это следствие 1 закона).
Доказательство справедливости закона для био систем: 1780 Лавуазье и Лаплас опыт с морскими свинками. Е хим. связей в белках, жирах и углеводах переходит в тепловую – метод непрямой (?) калориметрии. Свинок кормили – мерили тепло, столько же хавчика сжигали – тоже мерили, сравнили, получили числа одного порядка. По умному: совпадение тепловых эффектов при прямом сжигании продуктов и при их окислении в орг-ме морской свинки свид-т о том, что пути превращения прод-в питания в метаб-ких процессах и хим. р-циях вне живой клетки яв-ся эквивалент-ми с точки зрения суммарных тепловых эффектов. Живые орг-мы не являются источником новой Е. Окисление поступающих в живой организм пит. в-в приводит к высвобождению в нем эквивалетного к-ва Е.
Еще док-во: работа мышцы при 250С (289К = Т), КПД = 30% = 1/3.
Т1 = (298*3) /2 = 447К = 1740С.
Метод прямой калориметрии исполь-ся и на человеке (Этуотер). Исполь-ся герметичная камера: ч/з систему труб подается определ. кол-во О2, считают сколько выд-ся СО2, Н2О и т.д. есть датчики на t. Ограничения: 1) живой объект не должен накапливать массу и расти. 2) жив организм не должен совершать работу (физ. нагрузку). Кол-во Е, поглощенной за сутки чел. орг-мом вместе с пит. в-ми, равно выделенной за это же время теплоте. След-но, закон справедлив для жив. орг-мов.
Метод непрямой калориметрии: С полным и неполным газовым анализом. Ввели коэффициент: при расщеплении 1 мол-лы глюкозы исп-ся 6 мол-л О2, а выделяется 6 мол-л СО2, 6 мол-л Н2О и 678 кал. ДК = выд СО2 в ед времени / погл О2 в ед времени. Производят сравнение состава и объема вдых. и выдых. воздуха. Исп. мешок Дугласа. Для анализа исп. газоанализаторы: ГА Холдейна: система стеклян. трубочек, погл-щая CO2 и O2. Сейчас ГА с поглощением световых потоков. Нормальный дых. коэфф. 0,85±0,03. Нахождение КЭК (калориметрический эквивалент кислорода) – численно равен кол-ву Е, высвобождающейся в организме при потреблении 1 л О2. В клинических условиях исп. неполный ГА, не считают СО2. Считают объем поглощенного О2 с пом. спирографа. Диаграмма под наклоном, из замкнутой системы постепенно уходит О2.
10. Закон Гесса, его применимость к биопроцессам.Следствие закона Гесса, его практическое значение.
Теплоту у теплокровных раздел. на 2 типа:1) первичная (осн. обмен, Q, кот. выд-ся сразу после поглощения пищи при взаимод. с О2, т.е выд-ся в рез-те протекания биохим. проц-ов. В живых орг-мах любая хим. р-ция идет с КПД термин «тепловой эффект р-ции» заменяют термином «энтальпия р-ции». Энтальпия р-ции – это изменение энтальпии системы при протекании химической реакции. Она может быть больше нуля или меньше нуля. Если ∆H > 0, то Q > 0 (эндотермические реакции).
Для равновесного сост. S стремится к мах, U=0. Стац. сост. отличается тем, что S ≠ мах, а является постоянной величиной, S=const, U не равняется 0, U=const. Ежесекундный прирост энтропии стремится к min. Любая живая система может находиться только в стац. сост. Если достигнуто состояние ТД равновесия — это уже не живая система. Качество стационарного состояния может быть различным.
В открытых системах:
S состоит из двух показателей.
Si – внутри самой сист., S — самой системы, Se – внешняя среда.
dS=dSi+dSe (d – это ∆ — это изменение)
Когда dSe > dSi и dSe 0.
Состоянию ТД равновесия — характерно мах значение S (S=max), U=0, т.е. Е, которая расходуется на совершение А.
Сходство: стац. и равновесное состояния не зависят от времени.
Отличия стац. сост. от равновесия (из конспекта):
1) своб. Е (∆G) в стац. сост. есть величина постоянная во времени и не равна 0. В ТД равн. ∆G=const, но ∆G =0 => открытые сист., если вывести из стац. сост. могут совершать работу; при ТД равновесии не способны совершать работу.
2) энтропия. В стац. сост. =const, но она не max. (∆G) ∆S ≠ max = const.
3) . в стац. сост. проявляется кинетический параметр (фактор) (изменение энтропии во времени) dS/dt = dSi/dt + dSe/dt.
* постоянный обмен энергией с окружающей средой
* постоянно тратится свободная энергия на поддержание состояния
* т/д потенциалы постоянны, G и F не равны 0
* энтропия постоянна, но не максимальна
* отсутствует поток вещества и энергии в окружающую среду и обратно
* на поддержание этого состояния не затрачивается свободная энергия
* работа способности системы равна 0, т/д потенциалы равны 0
* в системе отсутствуют градиенты
Переход на новый стац. уровень:
2 пути: 1) «овершот» — по нему переходят живые организмы при изм внеш. усл. (приспособление). График.
Нижняя стрелочка – это старый стац. уровень.
Верхняя стрелочка – это новый стац. уровень.
2) «ложный старт» — усиление или уменьшение О2, выращивание лука с О2 и без. График. С О2 – аэробный распад углеродов. Без О2 – обмен в-в переходит на анаэробный путь. А если потом снова дать О2 – то получится график 2 (то что обведено кружочком – там осущ-ся уничтожение продуктов анаэробного пути). Пример для чела: пока не расщепится молочная к-та осуществлять работу дальше нельзя.
17. Теорема Пригожина и направленность эволюции биосистем. Энтропия и биологический прогресс.
Стац. сост. хар-ся min ежесекундным приростом энтропии (благодаря этому происходит эволюция).
Теорема: при постоянных внеш. усл. в системе, находящейся вблизи положения ТД равновесия в стац. сост., скорость возрастания энтропии, за счёт необходимости внутр. процессов, принимает постоянное минимальное значение отличное от нуля.
Или: В стационарных состояниях при фиксированных внешних параметрах локальная продукция энтропии в открытой т/д системе стремится к минимальному значению.
Энтропия – мера рассеивания свободной энергии, следовательно любая открытая т/д система в стационарном состоянии стремится к минимальному рассеиванию свободной энергии. Если в силу причин система отклонилась от стационарного состояния, то вследствие стремления системы к минимальной энтропии, в ней возникают внутренние изменения, возвращающие ее в стационарное состояние.
Величина, кот это всё характеризует:
β= T* (dS/dt), где β – диссипативная фукнкция. β>0, min. С этим связан Критерий эволюции открытых систем: ∆β/dt происходит преобразование одного вида Е в другой – это центральное событие фотосинтеза.
Физ-хим. сущность фотосинтеза: Ф. – процесс преобразования электромагнитной Е в Е хим. связей, сопровождающийся ↑ энергетического потенциала системы. Система является ТД открытой. При поглощении солнечного излучения растениями, водорослями, нек. микро, возрастают уровни свободной Е (∆F) и общей энергии (∆U), в которой значительную часть составляет Е электрона: ∆F = ∆U — Т∆S
КПД процесса фотосинтеза составляет обычно 6-8%, у хлореллы он достигает 20-25%. Большая часть поглощённой листом энергии теряется на тепловое излучение. В энергию химич. связей включается в ср. 1—2% поглощённой ФАР. Осн. показатель Ф.— его интенсивность, т. е. кол-во газа, поглощённого или выделенного единицей массы или поверхности листа в единицу времени. Интенсивность Ф. зависит от вида растений, состояния листьев, внеш. условий (свет, СO2). Ф. лесных древесных растений в 5—8 раз ниже, чем Ф. травянистых растений открытых местообитаний.
Реакции, протекающие под воздействием светового излучения, называются фотохимическими Основной закон фотохимии – закон квантовой эквивалентности (А. Эйнштейн, 1912 г): каждый поглощенный квант света hν вызывает изменение одной молекулы.
Важнейшим параметром фотохимической реакции служит квантовый выход γ:
γ = число фотохим. превращений/число поглощений квантов.
В зависимости от типа фотохимической реакции квантовый выход может меняться в широких пределах. Это связано с возможностью потери поглощенной энергии до фотопревращения. Если время существования фотовозбужденной молекулы и скорость фотодиссоциации совпадают, то γ
1. При γ >> 1 фотореакция идет по цепному механизму. В частности для реакции H2 + Cl2 = 2HCl γ = 105.
Типы фотохимических реакций:
1) Фотодиссоциация (фотолиз) приводит к разложению исходного вещества, поглотившего световую энергию. Примерами реакции фоторазложения служат такие: разложение галогенидов серебра (основа серебряной фотографии), фотолиз паров ацетона CH3CO CH3 → CO + другие продукты.
2) Фотосинтез приводит к образованию более сложных соединений. Примерами реакций фотосинтеза служат:
фотосинтез озона в верхних слоях атмосферы, создающий защитный озоновый слой:
О2→О +О — фотодиссоциация
О2+ О→ О3 — фотосинтез
фотосинтез органических соединений из углекислого газа, воды, минеральных веществ зелеными растениями. В частности, синтез глюкозы может быть описан уравнением:
6СО2 + 6Н2О →глюкоза + 6О2
3) Фотохромизм – явление обратимого изменения пространственного или электронного строения молекул под действием света, сопровождающееся изменением окраски вещества. На основе фотохромных материалов изготовляются линзы с переменным светопропусканием, оконные стекла, фотохромные системы на основе некоторых органических и координационных соединений.
Вариант 2.
Графическая зависимость скорости реакции от субстрата, фермента и температуры
42 Зависимость скор р-ции от темп. Ур Аррениуса
Для характеристики зависимости скорости химической реакции от температуры было введено понятие температурного коэффициента скорости (γ), равного отношению константы скорости при температуре (Т + 10) к константе скорости при температуре Т (т.е. γ показывает, во сколько раз изменяется константа скорости при увеличении температуры на 10 градусов):
Температурный коэф. Вант-гоффа-Q10= V t+10/V t.
Экспериментально было установлено, что повышение температуры на 10 К в области обычных температур (≈ 300 К) увеличивает скорость многих гомогенных реакций в 2 4 раза, т. е. для этих реакций γ = (2 4). Это правило называется правилом Вант–Гоффа.
В общем случае отношение констант скорости реакции k2 и k1, определенных при двух различных температурах Т2 и Т1, равно
Более точную зависимость скорости реакции от температуры дает уравнение Аррениуса:
где k — константа скорости реакции, R — универсальная газовая постоянная, Е — энергия активации химической реакции. В случае простых реакций величина Е показывает, какой минимальной (избыточной по сравнению со средней) энергией в расчете на 1 моль должны обладать реагирующие частицы, чтобы они могли вступить в химическую реакцию. В случае сложных реакций величина Е называется эмпирической или кажущейся энергией активации и в общем случае зависит от энергий активации отдельных стадий данной реакции.
Проинтегрировав уравнение (10.16), получим уравнение Аррениуса в интегральной форме:
где А — предэкспоненциальный множитель. Физический смысл А в случае простых реакций: мономолекулярных — это частота колебаний по разрываемой связи (А 1013 сек-1), бимолекулярных — величина А пропорциональна общему числу столкновений между молекулами реагирующих веществ (А 10-10 10-11 см3/(мол-л•сек).
Есть еще такое Ур-е: V= pZeEакт/RT, где р-стерический фактор, Z- число столкновений молекул, Еакт — энергия активации
Зависимость скорости реакции от температуры дает уравнение Аррениуса:
где k — константа скорости реакции, R — универсальная газовая постоянная, Е — энергия активации химической реакции. В случае простых реакций величина Е показывает, какой минимальной (избыточной по сравнению со средней) энергией в расчете на 1 моль должны обладать реагирующие частицы, чтобы они могли вступить в химическую реакцию. В случае сложных реакций величина Е называется эмпирической или кажущейся энергией активации и в общем случае зависит от энергий активации отдельных стадий данной реакции.
Проинтегрировав уравнение (10.16), получим уравнение Аррениуса в интегральной форме:
где А — предэкспоненциальный множитель. Физический смысл А в случае простых реакций: мономолекулярных — это частота колебаний по разрываемой связи (А 1013 сек-1), бимолекулярных — величина А пропорциональна общему числу столкновений между молекулами реагирующих веществ (А 10-10 10-11 см3/(мол-л•сек).
Проинтегрировав ур-е (10.16) в пределах температур от Т1 до Т2, получим:
Энергию активации можно определить как аналитически по уравнению (10.18), так и графическим методом. Для этого необходимо знать ряд констант скоростей при разных температурах. Если реакция подчиняется уравнению Аррениуса, то зависимость lnk от 1/T должна выражаться прямой линией, что следует из уравнения (10.17) (рис. 7).
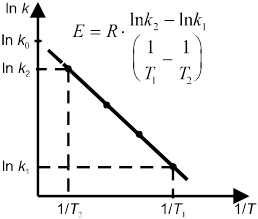
Для очень большого числа реакций энергия активации находится в пределах от 60 до 240 кДж/моль, т.е. примерно соответствует энергиям химических связей.
Энерг актив. Связана с Q10: Е=0,46T1*T2lgQ10
44 КИНЕТИКА ФЕРМЕНТ РЕАКЦИЙ, акт ферм, ед измерен акт и колич ферм.
ферм– это специфические органические катализаторы, синтезируемые живыми клетками. Как правило все ферменты представляют собой белки с различными молекулярными массами: от 9 кДа до 1000 кДа. Каждый фермент катализирует определённую химическую реакцию.
Субст- это вещества, с которыми происходит химическое превращение под действием ферментов. Субстратами ферментов могут быть как природные, так и химически синтезированные вещества.
1. с 1 активным центром (связывает S, расщепляет связи + связан с активацией и
2. кроме активного каталитического центра имеется аллостерический центр.
По взаимодействию с кофакторами:
1. ковалентная связь с кофактором — простетическая группа
2. нековалентное связывание — отделяется при гидролизе.
1. ускоряют протекание реакций
2. являются специфичными (мало-, относительно, абсолютно)
При присоединении субстрата к ферменту, его связи переходят в напряжённое состояние, что снижает энергию активации.
Факторы влияющие на активность:
t — идет как ферментативный процесс, так и денатурация белка. Q10 – 2

2. кислотность среды. Амилаза слюны — рН =7-7.5 , пепсин желудка
3. концентрация S
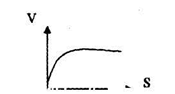
На начальном участке графика [S] низкая→ реакция первого порядка.
На втором — переходная стадия → переходный порядок реакции. На третьем участке (плато) → перенасыщение S→0 порядок реакции.
Кат. акт ферм — это способность фермента превращать большое количество молекул субстрата, в то время как сам он к концу реакции остается неизменным. Ферменты различаются по своей каталитической способности. Например, 1 моль трипсина осуществляет 102 циклов в секунду, глюкозоксидаза — 17×103 циклов в секунду и т.д. Это называется “числом оборотов фермента”. Число оборотов варьирует от 1 до 106 в зависимости от природы фермента. Каталитическую активность ферментов выражают в единицах активности.
Международная ед акт (E) — это количество фермента, которое катализирует превращение 1 мкмоля субстрата в 1 минуту в оптимальных условиях.
Ед акт в системе СИ (катал) — соответствует количеству фермента, которое катализирует превращение 1 моля субстрата в 1 секунду.
Соотношение между единицами активности: 1 E = 16,67 нанокатал; В медицине активность ферментов выражают чаще всего в единицах активности на 1 л биологической жидкости. Удельная активность фермента — это активность, выраженная в единицах активности на 1 мг (или 1 г) белка или 1 мг (1 г) препарата фермента. Используется в биохимической практике.
45 Осн положен теор ферм кинетики и общ теор действ фер-та
Предварительные эксперименты по изучению кинетики ферментативных реакций показали, что скорость реакции E + S —> E + P, вопреки теоретическим ожиданиям, не зависит от концентрации фермента и субстрата так, как в случае обычной реакции второго порядка. Самая ранняя попытка математически описать ферментативные реакции была предпринята Дюкло в 1898 г. Браун (1902) и независимо от него Анри (1903) впервые выдвинули гипотезу об образовании в ходе реакции фермент-субстратного комплекса. Это предположение основывалось на трех экспериментальных фактах:
1. папаин образовывал нерастворимое соединение с фибрином
2. субстрат инвертазы — сахароза могла защищать фермент от тепловой денатурации
3. было показано, что ферменты являются стереохимически специфическими катализаторами
Х-КА ДЕЙСТВИЯ ФЕРМ:
1. специфичность действия-способность ускорять протекание 1 или нескольких реакций (амилазная реакция расщепляет крахмал до глюкозы)
а. абсолютная- определенный субстрат;
б. относительная- ферменты, которые катализируют ращепление определенного типа связи. (пепсин)
2. ускорение протекания ферментативн. Реакций- каталитичность. Ферменты действуют в мыгких условиях (норм давление, pH, температура): гидролиз крахмала
Амилаза = 37 градусов, pH7, скорость выше, чем при неорган.катализе.
2. регулируемость – есть факторы под воздействием которых скорость может увеличится или уменшаться
В 1913 году Михаэлис и Ментен опубликовали свою теорию общего механизма ферментативных реакций. Их уравнение стало фундаментальным принципом всех кинетических исследований ферментов вот уже почти целый век.
т.е. фермент Е вступает во взаимодействие с субстратом S с образованием промежуточного комплекса ES, который далее распадается на свободный фермент и продукт реакции Р. Математическая обработка на основе закона действующих масс дала возможность вывести уравнение, названное в честь авторов уравнением Михаэлиса–Ментен, выражающее количественное соотношение между концентрацией субстрата и скоростью ферментативной реакции:
где v – наблюдаемая скорость реакции при данной концентрации субстрата [S]; KS– константа диссоциации фермент-субстратного комплекса, моль/л; Vmax– максимальная скорость реакции при полном насыщении фермента субстратом.
Основной механизм действия ферментов-они снижают Е активации за счет образования фермент-субстрат копмлекса.
Катализ приводит к ускорению достижения равновесия за счет снижения энергии активации (Еа), часто ступенчато.

Три стадии процесса:
1) E + S —— ES (K = k1/k-1) (БЫСТРАЯ)
2) ES —— EP (k2)(медленная)
Таким образом, в момент равновесия скорости образования и исчезновения энзимсубстратного комплекса (ES) равны:
Уравнение Михаэлиса — Ментен
Диаграмvа скорости реакции V как функции от концентрации [S].
Уравне́ние Михаэ́лиса — Ме́нтен — основное уравнение ферментативной кинетики, описывает зависимость скорости реакции, катализируемой ферментом, от концентрации субстрата и фермента. Простейшая кинетическая схема, для которой справедливо уравнение Михаэлиса:
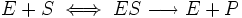
Уравнение имеет вид:
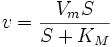 ,
,
· 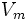 — максимальная скорость реакции, равная
— максимальная скорость реакции, равная 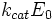 ;
;
· 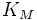 — константа Михаэлиса, равная концентрации субстрата, при которой скорость реакции составляет половину от максимальной;
— константа Михаэлиса, равная концентрации субстрата, при которой скорость реакции составляет половину от максимальной;
· 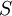 — концентрация субстрата.
— концентрация субстрата.
Вывод уравнения
Вывод уравнения был впервые предложен Бриггсом и Холдейном. Вывод уравнения скорости ферментативной реакции, описываемой схемой Михаэлиса-Ментен.
Обозначения констант скоростей:
k1 — константа скорости реакции образования фермент-субстратного комплекса из фермента и субстрата
k-1 — константа скорости реакции диссоциации фермент-субстратного комплекса на фермент и субстрат
k2 — константа скорости реакции превращения фермент-субстратного комплекса в фермент и продукт
Для фермент-субстратного комплекса применим метод квазистационарности, так как в подавляющем большинстве реакций константа скорости превращения фермент-субстратного комплекса в фермент и продукт много больше, чем константа скорости образования ферменто-субстратного комплекса из фермента и субстрата. Иными словами:
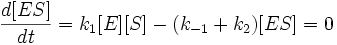
Учтем тот факт, что фермент, изначально находившийся только в свободной форме, в процессе реакции находится как в виде фермент-субстратного комплекса, так и в виде молекул свободного фермента. Таким образом:
Преобразуем это к виду:
И подставим в первое уравнение. После раскрытия скобок и группировки слагаемых получим следующее:
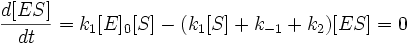
Выразим отсюда концентрацию фермент-субстратного комплекса:
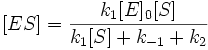
Скорость ферментативной реакции в целом (то есть скорость образования продукта) представляет собой скорость распада фермент-субстратного комплекса по реакции первого порядка с константой k2:
Подставим в эту формулу выражение, которое мы получили для концентрации ES. Получим:
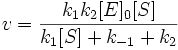
Разделим числитель и знаменатель на k1. В результате:
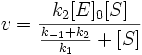
Выражение в знаменателе — (k-1+k2)/k1 — называется константой Михаэлиса (Km). Это кинетическая константа (с размерностью концентрации), которая равняется такой концентрации субстрата, при которой скорость ферментативной реакции составляет половину от максимального значения.
Для начальной стадии реакции можно пренебречь уменьшением концентрации субстрата. Тогда выражение для начальной скорости реакции будет выглядеть так:
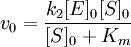
Если k-1>k2, то на первой стадии ферментативной реакции с течением времени устанавливается равновесие (квазиравновесный режим протекания реакции), и в выражение для скорости ферментативной реакции входит уже не константа Михаэлиса, а субстратная константа KS, характеризующая взаимодействие фермента с субстратом в равновесных условиях:
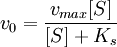 ;
; 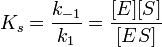
По значению KS можно судить о химическом сродстве субстрата к ферменту.
Металлоферменты и ферменты, активируемые металлами
Металлоферменты содержат определенное количество ионов металлов, имеющих функциональное значение и остающихся связанными с молекулой фермента в ходе его очистки. Ферменты, активируемые металлами, связывают последние менее прочно, но для своей активности требуют добавления металлов в среду. Таким образом, разграничение между металлоферментами и ферментами, активируемыми металлами, основано на сродстве данного фермента к иону «своего» металла. Механизмы, основанные на участии ионов металлов в катализе, в обоих случаях, по-видимому, сходны.
Тройные комплексы фермент—металл — субстрат
Для тройных (трехкомпонентных) комплексов, включающих каталитический центр (Enz), ион металла (М) и субстрат (S) со стехиометрией 1:1:1, возможны четыре различных схемы образования:
Enz—S—М М —Enz—S
Комплекс с мостиковым Комплекс с мостиковым субстратом ферментом
М
Enz—M—S Enz|
S
Простой комплекс с Циклический комплекс
мостиковым металлом с мостиковым металлом
В случае ферментов, активируемых металлами, реализуются все четыре схемы. Для металлофермен-тов образование комплекса Enz—S—М невозможно, иначе они не могли бы удерживать металл в процессе очистки (они находятся в форме Enz — — М). Можно сформулировать три общих правила.
1. Большинство (но не все) киназ (АТР:фосфо-трансферазы) образуют комплексы с мостиковым субстратом типа Enz—нуклеозид—М.
2. Фосфотрансферазы, использующие в качестве субстрата пируват или фосфоенолпируват, другие ферменты, катализирующие реакции с участием фосфоенолпирувата, а также карбоксилазы образуют комплексы с мостиковым металлом.
3. Данный фермент может быть способен к образованию мостикового комплекса одного типа с одним субстратом и другого типа — с другим.
Комплексы с мостиковым ферментом(М —Enz—S)
Металлы в комплексах с мостиковым ферментом, по-видимому, выполняют структурную роль, поддерживая активную конформацию (примером служит глутаминсинтаза), или образуют мостик с другим субстратом (как в пируваткиназе). В пиру-ваткиназе ион металла играет не только структурную роль, но и удерживает один из субстратов (АТР) и активирует его:
А
Пируваткиназа—АТР
Креатин.
Комплексы с мостиковым субстратом
Образование тройных комплексов с мостиковым субстратом, которое наблюдается при взаимодействии ферментов с нуклеозидтрифосфатами, по-видимому, связано с выстеснением Н20 из координационной сферы металла, место которой занимает АТР:
АТР- + M(H20) 2+ fi г± АТР—М(Н20) 2 \ + ЗН20.
Затем субстрат связывается с ферментом, образуя тройной комплекс:
АТР—М(Н20) 2 :, + Enz 2 :v
В фосфотрансферазных реакциях ионы металлов, как полагают, активируют атомы фосфора и образуют жесткий полифосфат-адениновый комплекс в соответствующей конформации, который включается в состав активного четырехкомпонентного комплекса.
Комплексы с мостиковым металлом
м
Enz-M-SnnnEnz |
S
Кристаллографические данные, а также анализ первичной структуры показывают, что в активных центрах многих белков в связывании металла участвует остаток гистидина (примерами служат кар-боксипептидаза А, цитохром с, рубредоксин, мет-миоглобин и метгемоглобин; см. гл. 6). Лимитирующей стадией образования бинарных (двухкомпо-нентных) комплексов Enz—М во многих случаях является вытеснение воды из координационной сферы иона металла. Активация многих пептидаз ионами металла является медленным процессом, длящимся несколько часов. Среди таких металлоферментов особое значение в живых системах имеют цитохромы и гемсодержащие ферменты, активными группировками которых служат железопорфириновые комплексы ( см. стр. [1]
Во многих металлоферментах, особенно катализирующих окислительно-восстановительные реакции, присутствуют именно металлы с переменной валентностью, действующие как акцепторы водорода, например железо, медь, марганец, молибден. Известно, что для активации таких ферментов, как ксантиноксидаза или нитроредуктаза, молибден должен быть в шестивалентном состоянии. [2]
Карбоксипептидаза — это металлофермент, содержащий один атом цинка на молекулу белка. Карбоксипептидаза катализирует гидролиз С-концевой пептидной связи в белках и олигопеп-тидах и сложных эфиров а-оксикислот. По данным рентгеноструктурного анализа Карбоксипептидаза представляет собой глобулярный белок, в котором содержится один атом цинка, координированный двумя остатками гистидина. Кроме того, в состав активного центра входят карбоксильная ( Glu-270), фенольная ( Туг-248) и гуанидиновая ( Arg-145) группы. [3]
Ингибирует широкий набор металлоферментов, включая гидроге-назу и гемоглобины ( при низких конц. СО-инги-бирование, обратимое под действием света, характерно для цитохромокси-дазы и др. гемсодержащих белков. Карбонилы Си и др. металлов не чувствительны к свету. Моноксид углерода сильно токсичен для многих живых тканей и микроорганизмов. [4]
СОД относится к металлоферментам, у которых в активном центре происходит восстановление и окисление иона металла. [5]
 | Трехмерная структура химотрипсина. |
Карбоксипептидаза А — это металлофермент, который, будучи экзопепти-дазой, гидролитически отщепляет С-концевые, прежде всего ароматические, остатки аминокислот. Для работы фермента необходим ион Zn2, который с некоторыми ограничениями может быть замещен на ионы других переходных металлов. [6]
Сравнительное изучение каталитических свойств различных металлоферментов в начале было главным образом ограничено оценкой их относительной каталитической активности. Однако детальное изучение катализа и связывания лиганда для серии металлоферментов может пролить свет на роль иона металла, если при отсутствии каталитической активности сохраняется связывание металла. [7]
Прочность связи фосфата с металлоферментом удивительно велика. Константа устойчивости комплекса, равная около 106 М 1 [51, 63, 76], выше, чем можно было бы ожидать для взаимодействия НРО f — и иона двухвалентного металла. Вероятно, вблизи иона цинка находится еще один катионный центр связывания, например протон аминогруппы боковой цепи аминокислоты, взаимодействующий с кислородными атомами фосфата. Образование фосфорилфермента при реакции с субстратом и НРО можно считать твердо установленным. После разрушения белка фосфат оказывается присоединенным к серину. Отсюда делается вывод о том, что в активном центре вблизи иона цинка находится остаток серина, ориентация которого делает возможной его атаку по фосфорильному атому присоединенного производного фосфата. Вполне вероятно, что фосфорильная группа, присоединенная к серину, сохраняет связь с ионом цинка. [8]
ЭКВ отчетливо проявляются в свойствах металлоферментов. Металлы служат кофакторами многих ферментов — в большинстве классов ферментов имеются металлозависимые. [9]
В части IV рассмотрены структуры металлоферментов и механизмы, посредством которых ионы металлов принимают участие в ферментативной активности, в частности, в разрыве связей. [10]
Малер [8] подчеркивает различия между металлоферментами, в которых металл прочно связан с белком ( по существу необратимо), и ферментами, активированными металлами, в которых связь металла с белком слаба и легкообратима. [11]
В настоящее время известно более 100 истинных металлоферментов, участвующих в большинстве реакций клеточного метаболизма. Многие из них передают электроны за счет металлов с переменной валентностью. В этом случае можно постулировать, что металл принимает непосредственное участие в осуществлении каталитического акта. [12]
Ферменты окислительно-восстановительного действия являются чаще всего металлоферментами, так как в качестве компонентов каталитически активных центров этих ферментов служат металлы. В истинных металлоферментах металл прочно связан в строго определенных стехиометрических соотношениях с теми или иными химическими группировками апофермента или со специальной группой небелковой природы. [13]
В этих же органах и тканях находятся соответствующие металлоферменты или ферменты, активируемые тем или иным металлом. Так, для меди, цинка, молибдена, селена, марганца критическим органом является печень; для марганца, кобальта — также и щитовидная железа; для кадмия и цинка — мужские половые железы и почки; для бериллия — костная ткань. Полагают также, что распределение металлов внутри клетки коррелирует с содержанием в ней металлосодержащих ферментов
Дата добавления: 2017-01-13 ; просмотров: 4681 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
http://poisk-ru.ru/s9637t3.html
http://helpiks.org/8-94053.html