Основным уравнение химической кинетики уравнение аррениуса
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Конспект лекций для студентов биофака ЮФУ (РГУ)
2.1 СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
2.1.9 Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Я. Г. Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ . Математически правило Вант-Гоффа можно записать следующим образом:
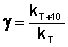 (II.29)
(II.29)
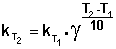 (II.30)
(II.30)
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
2.1.10 Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. С. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом:
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е’А выше, нежели энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
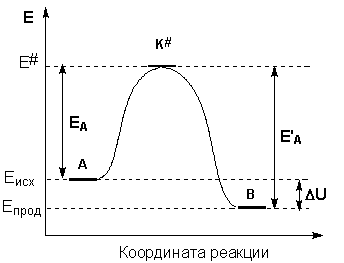
Рис. 2.5 Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.6):
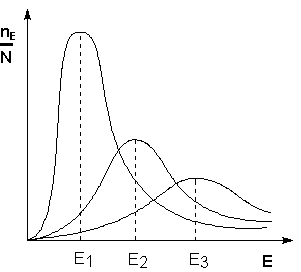
Рис. 2.6 Распределение частиц по энергии
Здесь nЕ/N – доля частиц, обладающих энергией E;
Ei — средняя энергия частиц при температуре Ti (T1 уравнения Аррениуса . Согласно уравнению изобары Вант-Гоффа,
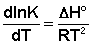 (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
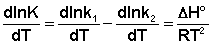 (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
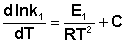 (II.33)
(II.33)
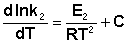 (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации :
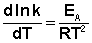 (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
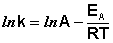 (II.36)
(II.36)
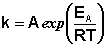 (II.37)
(II.37)
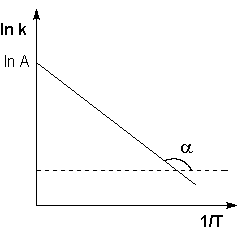
Рис. 2.7 Зависимость логарифма константы скорости химической
реакции от обратной температуры.
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
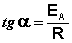 (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
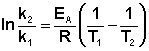 (II.39)
(II.39)
Copyright © С. И. Левченков, 1996 — 2005.
Несколько практических примеров использования уравнения Аррениуса.
1. На упаковке замороженного продукта написано, что его можно хранить на полке холодильника (5° С) в течение суток, в морозильнике, отмеченном одной звездочкой (-6° С), — неделю, двумя звездочками (-12° С) — месяц, а в морозильнике со значком *** (что означает температуру в нем -18° С) — 3 месяца. Предположив, что скорость порчи продукта обратно пропорциональна гарантийному сроку хранения tхр, в координатах lntхр, 1/Т получаем, в соответствии с уравнением Аррениуса, прямую. Из нее можно рассчитать энергию активации биохимических реакций, приводящие к порче данного продукта (около 115 кДж/моль). Из того же графика можно выяснить, до какой температуры надо охладить продукт, чтобы его можно было хранить, например, 3 года; получается -29° С.
2. Альпинисты знают, что в горах трудно сварить яйцо, и вообще любую пищу, требующую более или менее длительного кипячения. Качественно причина этого понятна: с понижением атмосферного давления уменьшается температура кипения воды. С помощью уравнения Аррениуса можно рассчитать, сколько времени потребуется, например, чтобы сварить вкрутую яйцо в г. Мехико, расположенном на высоте 2265 м, где нормальным считается давление 580 мм рт.ст., а вода при таком пониженном давлении кипит при 93° С. Энергия активации реакция «свертывания» (денатурации) белка была измерена и оказалась очень большой по сравнению со многими другими химическими реакциями — порядка 400 кДж/моль (она может несколько отличаться для различных белков). В таком случае понижение температуры от 100 до 93° С (то есть от 373 до 366 К) приведет к замедлению реакции в 10(400000/19)(1/366 — 1/373) = 11,8 раза. Именно поэтому жители высокогорья предпочитают варке пищи ее жарку: температура сковородки, в отличие от температуры кастрюли с кипятком, не зависит от атмосферного давления.
3. В кастрюле-скороварке пища готовится при повышенном давлении и, следовательно, при повышенной температуре кипения воды. Известно, что в обычной кастрюле говядина варится 2-3 часа, а компот из яблок — 10-15 мин. Учитывая, что оба процесса имеют близкую энергию активации (около 120 кДж/моль), можно по уравнению Аррениуса рассчитать, что в скороварке при 118°С мясо будет вариться 25-30 мин, а компот — всего 2 мин.
Уравнение Аррениуса очень важно для химической промышленности. При протекании экзотермической реакции выделяющаяся тепловая энергия нагревает не только окружающую среду, но и сами реагенты. это может привести к нежелательному сильному ускорению реакции. Расчет изменения скорости реакции и скорости тепловыделения при повышении температуры позволяет избежать теплового взрыва (см. ВЗРЫВЧАТЫЕ ВЕЩЕСТВА).
Зависимость скорости реакции от концентрации реагентов.
Скорость большинства реакций со временем постепенно снижается. Этот результат хорошо согласуется с теорией столкновений: по мере протекания реакции концентрации исходных веществ падают, снижается и частота столкновений между ними; соответственно уменьшается и частота столкновения активных молекул. Это приводит к уменьшению скорости реакции. В этом состоит сущность одного из основных законов химической кинетики: скорость химической реакции пропорциональна концентрации реагирующих молекул. Математически это можно записать в виде формулы
v = k[A][B], где k – постоянная, называемая константой скорости реакции. Приведенное уравнение называется уравнением скорости химической реакции или кинетическим уравнением.
Константа скорости для данной реакции не зависит от концентрации реагентов и от времени, но она зависит от температуры в соответствии с уравнением Аррениуса:
Простейшее уравнение скорости v = k[A][B]
всегда справедливо в том случае, когда молекулы (или другие частицы, например, ионы) А, сталкиваясь с молекулами В, могут непосредственно превращаться в продукты реакции. Подобные реакции, идущие в один прием (как говорят химики, в одну стадию), называются элементарными реакциями. Таких реакций немного. Большинство реакций (даже таких с виду таких простых как H2 + I2 = 2HI) не являются элементарными, поэтому исходя из стехиометрического уравнения такой реакции записать его кинетическое уравнение нельзя.
Кинетическое уравнение можно получить двумя способами:
— измеряя зависимость скорости реакции от концентрации каждого реагента по отдельности, и теоретически – если известен детальный механизм реакции. Чаще всего (но не всегда) кинетическое уравнение имеет вид
v = k[A]x[B]y, где x и y называются порядками реакции по реагентам А и В. Эти порядки, в общем случае, могут быть целыми и дробными, положительными и даже отрицательными. Например, кинетическое уравнение для реакции термического распада ацетальдегида
CH3CHO = CH4 + CO имеет вид
v = k[CH3CHO]1,5, т.е. реакция имеет полуторный порядок. Иногда возможно случайное совпадение стехиометрических коэффициентов и порядков реакции. Так, эксперимент показывает, что реакция
имеет первый порядок как по водороду, так и по иоду, то есть ее кинетическое уравнение имеет вид
v = k[H2][I2] (именно поэтому эту реакцию в течение многих десятилетий считали элементарной, пока в 1967 не был доказан ее более сложный механизм).
Если известно кинетическое уравнение, т.е. известно, как скорость реакции зависит от концентраций реагентов в каждый момент времени, и известна константа скорости, то можно рассчитать зависимость от времени концентраций реагентов и продуктов реакции, т.е. теоретически получить все кинетические кривые. Для таких расчетов используются методы высшей математики или компьютерные расчеты, и они не представляют принципиальных трудностей.
С другой стороны, полученное экспериментально кинетическое уравнение помогает судить о механизме реакции, т.е. о совокупности простых (элементарных) реакций. Выяснение механизмов реакций является важнейшей задачей химической кинетики. Это очень трудная задача, так как механизм даже простой с виду реакции может включать множество элементарных стадий.
История учения о химическом процессе. М., Наука, 1981
Леенсон И.А. Химические реакции. М., АСТ – Астрель, 2002
Теория активных столкновений
Теория активных столкновений (С. Аррениус) основана на том, что химическое взаимодействие осуществляется только при столкновении активных частиц, которые обладают достаточной энергией для преодоления потенциального барьера реакции и ориентированы в пространстве друг относительно друга. Чтобы произошла реакция, частицы в момент столкновения должны обладать некоторым минимальным избытком энергии, называемым энергией активации.
В теории активных столкновений считается, что акт превращения начальных веществ в конечные продукты совершается в момент столкновения активных молекул и протекает мгновенно. При этом молекулы рассматриваются как бесструктурные частицы, хотя в действительности химические реакции происходят путем постепенной перестройки молекул и перераспределения энергии между химическими связями.
Согласно молекулярно-кинетической теории энергия активации равна разности между средней энергии активных столкновений и средней энергии всех столкновений. Доля активных молекул, как показывают расчеты, составляет примерно от 10 -20 до 10 -10 . Если эта доля меньше, то скорость реакции мала, если же она больше, то реакция происходит быстро, иногда практически мгновенно. Чем выше энергия активации данной реакции, тем при более высоких температурах она совершается. Энергия активации ниже энергии диссоциации реагирующих молекул, так как для протекания реакции достаточно такого ослабления связей в молекулах, при котором начинают преобладать силы образования новых связей.
Источники активации могут быть самые разнообразные. Реакции между ионами в растворе происходят с небольшой энергией активации, которая требуется для дегидратации ионов. Реакции между свободными атомами и радикалами не требуют энергии активации, так как атомы и радикалы являются активными частицами. В гомогенных газовых реакциях основным источником активации служат столкновения, доля которых определяется законом распределения Больцмана и растет с температурой. В гетерогенных каталитических реакциях источниками активации могут служить изменения, происходящие в реагирующих молекулах при адсорбции их поверхностью катализатора. Активация может быть вызвана также внешними причинами: поглощением квантов света при фотохимических реакциях, действием электрических разрядов, ударом электронов, α – частиц, нейтронов и других излучений.
При подсчете числа столкновений нужно учитывать эффективный диаметр молекул ?. Рассмотрим элементарную бимолекулярную реакцию:
А + В → Продукты (XI.1)
Предположим, что молекула А неподвижна, а молекулы В движутся в пространстве параллельно прямой, проходящей через центр молекулы А. При отсутствии взаимодействия между молекулами А и В с молекулой А столкнутся все молекулы В, центры которых находятся внутри цилиндра, имеющего радиус r.

где σ1 и σ2 – диаметры молекул А и В соответственно. При притяжении между молекулами А и В прямолинейные пути молекул В, начиная с некоторого расстояния, искривляются, и молекулы сближаются, в результате чего с молекулой А столкнется часть молекул В, центры которых первоначально находились вне цилиндра с радиусом r. Тогда

При отталкивании молекул

где σ’1, σ’2, σ»1, σ»2 – эффективные диаметры молекул. Таким образом, эффективный диаметр молекул характеризует не только диаметры сталкивающихся молекул, но и взаимодействие между ними. Величина πσ 2 12 называется сечением соударений и имеет большое значение в современной теории кинетики химических реакций.
Эффективный диаметр σ молекул одного вида в газе рассчитывается с помощью молекулярно-кинетической теории или по эмпирическим уравнениям. Средний эффективный диаметр при столкновении с молекул разного вида вычисляем по уравнению
 (XI.2)
(XI.2)
Согласно молекулярно-кинетической теории газов полное число столкновений L0 за 1 с в 1 м 3 между одинаковыми молекулами рассчитываются по уравнению
 (XI.3)
(XI.3)
где n – число молекул в 1 м 3 ; m – масса частиц, кг.
Если в системе реагируют молекулы двух разных видов, то
 (XI.4)
(XI.4)
Число столкновений активных молекул La, рассчитанное на основе закона распределения Максвелла–Больцмана, определяется соотношением
 (XI.5)
(XI.5)
где L0 – полное число столкновений; E’ – энергия активации.
Исходя из теории активных соударений и молекулярно-кинетических представлений, вычислим константу скорости элементарной бимолекулярной реакции (XI.1) с участием молекул двух видов. Скорость рассматриваемой элементарной реакции согласно основному постулату химической кинетики выражается уравнением
где k – константа скорости; c1 и c2 – концентрации веществ А и В, моль/м 3 ;
 и
и  (XI.7)
(XI.7)
где n1 и n2 – число частиц А и В в 1 м 3 ; NA – число Авогадро.
Число активных столкновений равно числу реагирующих молекул А или В:
 (XI.8)
(XI.8)
При этом скорость реакции
 (XI.9)
(XI.9)
И с учетом (XI.8) и (XI.5) примет вид
 (XI.10)
(XI.10)
Приравнивая правые части уравнений (XI.6) и (XI.10) с учетом (XI.7), получаем
 (XI.11)
(XI.11)
Подставляя L0 из (XI.4) в (XI.11), получаем уравнение для расчета константы скорости реакции
 (XI.12)
(XI.12)
 (XI.13)
(XI.13)
 (XI.14)
(XI.14)
Можно написать вместо (XI.13)
Подставляя (XI.15) в (XI.12), получаем:
 (XI.16)
(XI.16)
Логарифмирование уравнения (XI.16) дает:
 (XI.17)
(XI.17)
Дифференцирование по T равенства (XI.17) приводит к соотношению (так как В’ приближенно не зависит от температуры)
 (XI.18)
(XI.18)
Прологарифмированное уравнение Аррениуса

где k – константа скорости реакции; A – предэкспоненциальный множитель; E – энергия активации. Продифференцируем его и сравним с (XI.18), то получим
 (XI.19)
(XI.19)
При температуре 300 — 400 К RT/2 = 1,2 — 1,4 кДж/моль. Поскольку энергия активации химической реакции обычно имеет значение от 50 до 200 кДж/моль, то при практических расчетах можно считать Е ≈ Е’. Поэтому для приближенного расчета констант скоростей бимолекулярных реакций вместо Е’ можно использовать энергию активации Е, вычисленную по уравнению Аррениуса на основании опытных данных.
Уравнение (XI.12) можно рассматривать как теоретическое обоснование уравнения Аррениуса на основе теории активных столкновений. Энергия активации в теории активных столкновений не вычисляется, а определяется опытным путем по зависимости скорости реакции от температуры. (Для некоторых сравнительно простых элементарных реакций энергия активации может быть вычислена из квантово-химических представлений). Предэкспоненциальные множители для бимолекулярных элементарных реакций рассчитываются по уравнению (XI.13). Однако бимолекулярные реакции, для которых экспериментально найденные предэкспоненциальные множители совпадают с рассчитанными, встречаются сравнительно редко. Чаще всего предэкспоненциальные множители, рассчитанные теоретически как для реакций в газах, так и растворах, значительно превышают экспериментальные значения. Это связано с упрощенным характером теории активных столкновений, которая считает, что столкновения между молекулами аналогичны столкновениям упругих шаров. В связи с этим в уравнение (XI.12) вводится множитель P, учитывающий отклонение теоретических расчетов от опытных данных. Этот множитель называется стерическим или энтропийным фактором. Уравнение (XI.12) с учетом этого фактора принимает вид
 (XI.20)
(XI.20)
При столкновении активных молекул должно быть вполне определенное расположение в пространстве активных групп, входящих в состав молекулы, которое бы обеспечило образование конечных продуктов. Стерический фактор P в большинстве случаев характеризует вероятность определенной геометрической конфигурации частиц при столкновении.
Есть и другие причины, приводящие к расхождению теории активных столкновений с опытом, которые также учитываются стерическим или энтропийным фактором. Вследствие туннельного эффекта элементарный акт может произойти при значениях энергии активации меньше Е. Это формально характеризуется величиной Р > 1. Вновь образующиеся молекулы могут быть сильно возбужденными. Если такие молекулы не освободятся от избытка энергии после своего возникновения, то они вновь могут превратиться в молекулы исходного вещества; в этом случае Р > k2
 (XI.94)
(XI.94)
где k1 = cAB / (cA cB) – константа равновесия. Вторая стадия реакции является лимитирующей.
Во втором предельном случае при k-1 3 раствора; dc2 / dr – градиент концентрации вещества B.
Чтобы определить поток J , разделим переменные r и c2 и проинтегрируем (XI.96) от R * до ∞ и от 0 до c2:
 (XI.97)
(XI.97)
где R * – расстояние между молекулами A и B при образовании пары столкновения; полагаем, что на расстоянии меньше R, c2 = 0.
Для простоты полагаем, что раствор достаточно разбавленный, и в процессе диффузии молекул B к молекуле A через слой раствора они не встречаются с другими молекулами, A и, поэтому, поток J имеет постоянное значение.
В результате интегрирования (XI.97), получаем

Скорость реакции, контролируемая диффузией, определяется скоростью потока молекул B к молекуле A. При этом скорость реакции w равна числу пар столкновения, которые образуются в 1 с в 1 м 3 раствора:
 (XI.99)
(XI.99)
где n1 = NA c1 – число молекул A в 1 м 3 раствора.
Подставляя значение J из (XI.98) в (XI.99), определяем
где D = D1 + D2, так как нужно учесть, что молекулы A также диффундируют в растворе навстречу молекулам B.
Приравнивая правые части уравнений (XI.100) и (XI.92), получаем для k3 выражение
Это выражение можно преобразовать, если для D1 и D2 использовать соотношение Стокса–Эйнштейна
 и
и  (XI.102)
(XI.102)
 (XI.103)
(XI.103)
где η – коэффициент вязкости раствора (или растворителя для достаточно разбавленного раствора). Подставляя (XI.103) в (XI.101), получаем
 ;
;  (XI.104)
(XI.104)
Таким образом, в рассмотренном предельном случае ( k1 ≠ → Продукты (XI.105)
константа скорости согласно (*) (при χ = 1) равна
 (XI.106)
(XI.106)
Но термодинамическая константа равновесия в растворе выражается через активности
 (XI.107)
(XI.107)
 (XI.108)
(XI.108)
Подставляя уравнение (XI.108) в (XI.106), получаем
 (XI.109)
(XI.109)
При γA = γB = γ ≠ = 1 уравнение (XI.109) преобразуется к виду

где k0 – константа скорости реакции, коэффициенты активности равны единице. Между константами k и k0 имеется соотношение
 (XI.110)
(XI.110)
С этим случаем мы встречаемся, например, когда одна и та же реакция может проводиться как в газовой фазе ( k0 ), так и в растворе ( k ).
Если исходные вещества A и B являются молекулами и, кроме того, принять γA = γB = γ ≠ = γ, то из выражения (XI.110) получаем, что k = k0 γ. Это означает, что для бимолекулярных реакций между молекулами константы скорости реакции при проведении ее в газовой фазе и в растворе различны.
Для мономолекулярной реакции
А → А ≠ → Продукты (XI.111)
Рассуждая аналогично, получаем выражение
 (XI.112)
(XI.112)
Если вещество A находится в растворе в молекулярной форме и принимая γA = γ ≠ = γ, то из уравнения (XI.112) получаем, что k = k0, т.е. для мономолекулярной реакции теория абсолютных скоростей реакций предсказывает слабое влияние растворителя и его природы на кинетику реакции, если конфигурация активированного комплекса мало отличается от исходных молекул.
Опыт во многих случаях подтверждает теорию. Например, мономолекулярная реакция разложения оксида азота ( N2O5 ) в газовой фазе при температуре 293 К имеет константу скорости, равную 3,4 ⋅ 10 -5 с -1 . При использовании в качестве растворителя хлороформа, дихлорэтана, нитрометана, жидкого брома и тетрахлорида углерода константы скорости при той же температуре равны соответственно 3,7 ⋅ 10 -5 ; 4,2 ⋅ 10 -5 ; 3,1 ⋅ 10 -5 ; 4,1 ⋅ 10 -5 с -1 . В случае бимолекулярных элементарных реакций перенос реакции из газовой фазы в раствор, а также изменение природы растворителя, как правило, заметно влияют на константу скорости реакции в соответствии с предсказанием теории абсолютных скоростей реакций.
Важное подтверждение теории абсолютных скоростей реакций получила для реакций между ионами в растворах сильных электролитов, так как в этом случае коэффициенты активности могут быть вычислены из теории сильных электролитов Дебая–Хюккеля. Если раствор электролита разбавленный, то коэффициенты активности можно выразить приближенно с помощью предельного закона Дебая–Хюккеля:
 (XI.113)
(XI.113)
где A – теоретический коэффициент, который для водных растворов равен 0,509; zi – заряд i-го иона; I – ионная сила раствора.
Если в реакции (XI.105) исходные вещества A и B являются ионами с зарядами zA и zB , то коэффициенты активности в уравнении (XI.110) можно приближенно выразить из уравнения (XI.113), принимая, что заряд активированного комплекса равен алгебраической сумме zA + zB зарядов реагирующих ионов:


 (XI.114)
(XI.114)

Таким образом, из теории активированного комплекса следует, что если в бимолекулярной реакции в растворе участвуют два иона с одинаковыми зарядами ( zA zB > 0 ), то lg ( k / k0 ) > 0 и константа скорости реакции увеличивается с ростом ионной силы раствора. Если же заряды ионов противоположные ( zA zB < 0 ), то и константа скорости реакции уменьшается с ростом ионной силы. Кроме того, согласно уравнению (XI.114) график в координатах lg ( k / k0 ) — √I в достаточно разбавленных растворах должен изображаться прямыми линиями, выходящими из начала координат, причем чем больше произведение zA zB, тем больше должен быть угол наклона этих прямых. Опытные данные хорошо подтверждают предсказания теории (рис.2).
Влияние ионной силы раствора на константу скорости реакции между ионами из-за изменения коэффициента активности ионов в растворах сильных электролитов называется первичным (или кинетическим) солевым эффектом. В реакциях с участием одного из ионов слабого электролита (например, иона водорода слабой кислоты) посторонний электролиз может влиять непосредственно на его концентрацию и, следовательно, на скорость реакции. Это – вторичный солевой эффект.
http://examchemistry.com/content/lesson/himreakcii/kinetika.html
http://www.corrosion.su/the_theory_of_active_collisions.php