Фундаментальные уравнения термодинамики. Преобразования Лежандра.
Глава 4. фундаментальные уравнения термодинамики. характеристические функции.
Фундаментальные уравнения термодинамики. Преобразования Лежандра.
Термодинамика как наука о наиболее общих свойствах макроскопических систем, находящихся в состоянии термодинамического равновесия, и о процессах перехода между этими состояниями построена просто – опытным путем установлены два ее основных закона, а применение к ним математического аппарата позволяет получить очень важные термодинамические соотношения.
Основой математического аппарата термодинамики является объединенное уравнение первого и второго законов термодинамики илифундаментальное уравнение Гиббса, которое для обратимых процессов записывается в виде
 , (4.1)
, (4.1)
где все параметры относятся к системе; PdV – механическая работа расширения системы; δW* – полезная работа системы (сумма немеханических видов работы, см. раздел 2.3)
Для простых систем в случае обратимых процессов фундаментальное уравнение записывается в виде
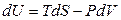 . (4.2)
. (4.2)
В математике независимыми переменными считаются те, которые стоят под знаком дифференциала. В фундаментальном уравнении (4.1) независимыми переменными являются S, V и все yk. Однако эти независимые параметры неудобны, так как энтропия непосредственно не измеряется, а объем легко определяется только для газов. Поэтому возникает задача перехода к новым независимым переменным.
Преобразование, меняющее ролями зависимые и независимые переменные, носит название преобразования Лежандра.
Рассмотрим функцию нескольких переменных:
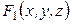 .
.
Полный дифференциал этой функции равен
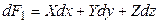 , (4.3)
, (4.3)
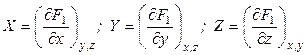 ,
,
Введем новую функцию
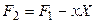 . (4.4)
. (4.4)
Полный дифференциал этой функции будет равен
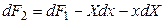 . (4.5)
. (4.5)
Подставив в равенство (4.5) значение dF1 из (4.3), получим:
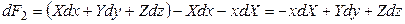 . (4.6)
. (4.6)
Cледовательно, в результате преобразования осуществлен переход от независимых переменных x, y, z к независимым переменным X, y, z, то есть переменная х стала зависимой, а Х – независимой. Кроме того, получили новую функцию F2. Таким образом, чтобы поменять зависимую переменную на независимую, следует воспользоваться соотношением
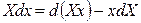 . (4.7)
. (4.7)
Впервые преобразование Лежандра к термодинамическим функциям применил Ф. Масье в 1869 году. Фундаментальное уравнение темодинамики для простых систем как для обратимых, так и для необратимых процессов запишется в виде:
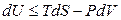 . (4.8)
. (4.8)
Знак неравенства используется для необратимых процессов, а знак равенства – для обратимых процессов. Применив к произведению PdV соотношение (4.7)
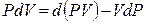 ,
,
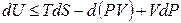 , (4.9)
, (4.9)
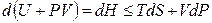 . (4.10)
. (4.10)
В результате перешли к независимым переменным S и Р и получили под знаком дифференциала в левой части (4.10) новую функцию
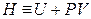 , (4.11)
, (4.11)
которая называется энтальпией.
Воспользовавшись подстановкой Лежандра для произведения TdS
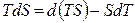 , (4.12)
, (4.12)
и подставив ее в фундаментальное уравнение (4.8), получим:
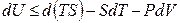 , (4.13)
, (4.13)
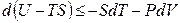 . (4.14)
. (4.14)
Стоящую под знаком полного дифференциала функцию U – TS обозначают по рекомендациям IUPAC символом Аи называютэнергией Гельмгольца(в некоторых учебниках энергию Гельмгольца до настоящего времени обозначают символом F):
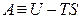 . (4.15)
. (4.15)
Итак, соотношение (4.14) записывается следующим образом:
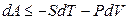 , (4.16)
, (4.16)
то есть в результате преобразований введена новая функция состояния при независимых переменных Т и V.
Преобразовав по Лежандру сразу оба произведения TdS и PdV в уравнении (4.8), получим:
 , (4.17)
, (4.17)
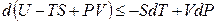 . (4.18)
. (4.18)
Функция U – TS + PV обозначается символом Gи называется энергией Гиббса. Следовательно, соотношение (4.18) запишется в виде:
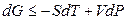 , (4.19)
, (4.19)
и энергия Гиббса является функцией независимых переменных Т и Р.
Дата добавления: 2015-08-20 ; просмотров: 3132 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Экзаменационный билет № 3
Для описания термодинамики поверхностных явлений применяют два метода: метод избыточных величин Гиббса и метод «слоя конечной толщины».
 Чтобы не определять границы поверхностного слоя (поверхности разрыва по Гиббсу), Гиббс предложил относить все изменения термодинамических параметров в слое в сравнении с параметрами объемной фазы к разделяющей поверхности, не имеющей объема или толщины (метод избыточных величин Гиббса). При таком рассмотрении поверхность характеризуется избыточными термодинамическими параметрами, непосредственно отражающими проявление поверхностной энергии. Объемные фазы считаются однородными вплоть до разделяющей поверхности. В соответствии с методом избыточных величин энергия Гиббса системы равна сумме энергий Гиббса G1 и G2 объемных фаз 1, 2 и поверхностной энергии Гиббса σs, которая является избыточной:
Чтобы не определять границы поверхностного слоя (поверхности разрыва по Гиббсу), Гиббс предложил относить все изменения термодинамических параметров в слое в сравнении с параметрами объемной фазы к разделяющей поверхности, не имеющей объема или толщины (метод избыточных величин Гиббса). При таком рассмотрении поверхность характеризуется избыточными термодинамическими параметрами, непосредственно отражающими проявление поверхностной энергии. Объемные фазы считаются однородными вплоть до разделяющей поверхности. В соответствии с методом избыточных величин энергия Гиббса системы равна сумме энергий Гиббса G1 и G2 объемных фаз 1, 2 и поверхностной энергии Гиббса σs, которая является избыточной: 
Характеристика величины адсорбции определяющася избытком вещества в поверхностном слое определенной толщины по сравнению с его количеством в таком же объеме фазы, также отнесенным к единице площади поверхности или единице массы адсорбента, называется гиббсовской адсорбцией и обозначается буквой гамма Г (метод избыточных величин Гиббса).
Объединенное уравнение первого и второго начал термодинамики для внутренней энергии поверхности с учетом поверхностной и химической энергии имеет вид (объем поверхностного слоя равен нулю)


Так как внутренняя энергия поверхности пропорциональна экстенсивным величинам, то
и ее полный дифференциал от тех же переменных запишется следующим образом:


Подставляя значение dU, получим:
 Для условия постоянства температуры это соотношение принимает вид
Для условия постоянства температуры это соотношение принимает вид

Разделив полученное уравнение на площадь поверхности, получим:
Данное уравнение называют фундаментальным адсорбционным уравнением Гиббса.
Для адсорбции одного конкретного вещества при постоянных химических потенциалах других веществ полученное уравнение можно записать относительно частной производной для данного компонента:

Принимая во внимание, что 
Для гиббсовской адсорбции это выражение принимает вид

Для растворенного вещества это выражение переходит в широко используемые адсорбционные уравнения Гиббса для неэлектролитов  | и для электролитов при их диссоциации в полярной фазе и отсутствии ее в поверхностном слое (при неполярной второй фазе):  |
Общее определение поверхностной активности дается соотношением:

которое справедливо как для неэлектролитов, так и для электролитов.
Поверхностная активность, как и гиббсовская адсорбция, может быть положительной и отрицательной. Абсолютное значение и ее знак зависят от природы как адсорбируемого вещества, так и среды (растворителя). Если с увеличением концентрации вещества поверхностное натяжение на границе раздела фаз понижается, то такое вещество называют поверхностно-активным. Для таких веществ 
Вещества, повышающие поверхностное натяжение на границе раздела фаз с увеличением концентрации, называют поверхностно-инактивными. Для них 
- Современная теория строения ДЭС (теория Штерна); роль специфической адсорбции, перезарядка поверхности. Примеры образования ДЭС. Строение мицеллы.
 Современная теория строения двойного электрического слоя основана на представлениях Штерна. Она объединяет две предыдущие теории. Согласно современной теории слой противоионов состоит из двух частей.
Современная теория строения двойного электрического слоя основана на представлениях Штерна. Она объединяет две предыдущие теории. Согласно современной теории слой противоионов состоит из двух частей.
Одна часть примыкает непосредственно к межфазной поверхности и образует адсорбционный слои (слой Гельмгсиьца) толщиной δ, которая равна радиусу гидратированных ионов, его составляющих. Другая часть противононов находится в диффузной части — диффузный слой (слой Гуи) с потенциалом φδ, толщина λ которой может быть значительной и зависит от свойств и состава системы. Потенциал в диффузной части двойного электрического слоя не может зависеть линейно от расстояния, так как ионы в нем распределены неравномерно. В соответствии с принятыми представлениями потенциал в слое Гельмгольца при увеличении расстояния от слоя потенциалопределяющих ионов снижается до потенциала диффузного слоя линейно, а затем, как будет показано, по экспоненте. Теория Штерна учитывает также специфическую (некулоновскую, химическую) составляющую адсорбции ионов на поверхности раздела фаз, которая существенным образом может влиять на изменение потенциала.
Пренебрежение размерами ионов приводит к тому, что не принимается во внимание толщина адсорбционною слон, и это, в свою очередь, вызывает большие погрешности при расчете параметров двойного электрического слоя. Кроме того, теория Гуи — Чепмена рассматривая только влияние концентрации и заряда ионов электролитов на изменение потенциала, не объясняет различного действия ионов разной природы, связанного со специфической адсорбцией их на межфазной поверхности.
Штерн предложил рассматривать слой противоионов состоящим из двух частей: внутренней (плотный слой Гельмгольца) и внешней (диффузный слой). Таким образом, теорию Гуи — Чепмена можно использовать для описания только строения внешней части слоя, где можно пренебречь адсорбционными силами и размерами ионов. Внутреннюю (плотную) часть Штерн представил как адсорбционный моноионный слой, в котором противоионы примыкают к поверхности благодаря электростатическим силам и специфическому взаимодействию. Введенный Штерном потенциал φδ часто называют штерновским. В плотной части двойного электрического слоя потенциал уменьшается линейно от φ0 до φδ. Принимая текущими переменными φ и х вместо φδ и δ, получим: 
Штерн попытался учесть влияние специфической адсорбции ионов на электрический потенциал, обусловленный действием ковалентных сил дополнительно к электростатическим силам. Так как радиус действия сил такой адсорбции соизмерим с размером ионов, это дает основание учитывать копалентные силы только для ионов, входящих в плотный слой Гельмгольца. Как видно из рисунка, плотность поверхностного заряда противоионов можно разделить на две части: плотность заряда qГ, обусловленного моноионным слоем, представляющим собой слой Гельмгольца, и плотность заряда qδ диффузного слоя Гуи. Общая поверхностная плотность заряда двойного электрического слоя равна сумме поверхностных плотностей зарядов плотного и диффузного слоев с обратным знаком: 
По Штерну, заряд слоя Гельмгольца складывается из заряда ионов, адсорбированых как за счет электростатического адсорбционного потенциала Fzφ, так и за счет потенциала
специфической адсорбции Ф. Было предположено, что поверхность имеет определенное число адсорбционных центров, каждый из которых взаимодействует с одним противоионом.
Пример образования ДЭС:
 Добавление в систему металл — вода раствора хлорида натрия приводит к избирательной адсорбции хлорид-анионов на поверхности металла. Появляется избыточный отрицательный заряд на поверхности металла и избыточный положительный заряд (ионы натрия) в близлежащем слое раствора, т. е. на межфазной поверхности образуется двойной электрический слой.
Добавление в систему металл — вода раствора хлорида натрия приводит к избирательной адсорбции хлорид-анионов на поверхности металла. Появляется избыточный отрицательный заряд на поверхности металла и избыточный положительный заряд (ионы натрия) в близлежащем слое раствора, т. е. на межфазной поверхности образуется двойной электрический слой.
Сильно адсорбирующиеся ионы в плотном слое иногда способны не только полностью скомпенсировать поверхностный потенциал, но и создать избыточный заряд со знаком заряда противоионов. Это явление называется перезарядкой. Перезарядка приводит к смене противоионов в диффузном слое на ионы с зарядом другого знака.
На рисунке видно, что при перезарядке поверхностный потенциал φ0 и потенциал диффузного слоя φδ имеют разные знаки.
 На формирование двойного электрического слоя существенное влияние оказывает природа поверхности конденсированной фазы, наличие определенных ионов в растворе, их концентрация. Рассмотрим систему водный раствор — поверхность иодида серебра. При избытке в растворе ионов серебра, например при добавлении нитрата серебра, эти ионы являются потенцналопределяюшими. В роли противоионов выступают нитрат-ионы, часть которых находится в плотном слое, а другая часть — в диффузном слое. Для такой системы формулу двойного электрического слоя можно записать следующим образом:
На формирование двойного электрического слоя существенное влияние оказывает природа поверхности конденсированной фазы, наличие определенных ионов в растворе, их концентрация. Рассмотрим систему водный раствор — поверхность иодида серебра. При избытке в растворе ионов серебра, например при добавлении нитрата серебра, эти ионы являются потенцналопределяюшими. В роли противоионов выступают нитрат-ионы, часть которых находится в плотном слое, а другая часть — в диффузном слое. Для такой системы формулу двойного электрического слоя можно записать следующим образом: 
В дисперсных системах двойной электрический слой возникает на поверхности частиц. Частицу дисперсной фазы в гетерогенно-дисперсной системе вместе с двойным электрическим слоем называют мицеллой. Строение мицеллы можно показать той же формулой, что и строение двойного электрического слоя. Внутреннюю часть мицеллы составляет агрегат основного вещества. На поверхности агрегата расположены потенциалопре-деляющие ионы. Агрегат вместе с потенциалопределяющими ионами составляет ядро мицеллы. Ядро с противоионами плотной части двойного электрического слоя образуют гранулу. Гранулу окружают противоионы диффузного слоя. Мицелла в отличие от гранулы электронейтральна.
Характеристические функции. Уравнения Гиббса-Гельмгольца
Функция называется характеристической, если с помощью этой функции и ее частных производных можно определить все термодина-мические свойства системы (p, V, T, S и др.)
Наиболее часто употребляемыми в химической термодинамике из характеристических функций U, H, F, Z являются энергия Гиббса (G) и энергия Гельмгольца (F).
Рассмотрим энергию Гиббса как функцию давления и температуры т.е. G = f (T, p).
Полный дифференциал функции двух переменных можно записать как сумму частных производных этой функции по независимым переменным, умноженных на дифференциалы независимых переменных, т.е.
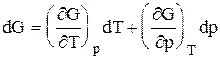 (6.126)
(6.126)
Но из уравнения (6.123)
Приравнивая коэффициенты при дифференциалах dT и dp в выражениях (6.123) и (6.126), получаем
 (6.127)
(6.127)
Эти выражения (6.127) показывают, что частные производные от G дают в простом и явном виде такие свойства системы как S и V.
Аналогично для свободной энергии Гельмгольца, если ее рассматривать как функцию объема и температуры F = f (V,T).

Сравнивая это выражение с уравнением (17), получаем
 (6.128)
(6.128)
Рассматривая энтальпию Н, как функцию давления и энтропии, т.е. H = f (p,S) и учитывая, что
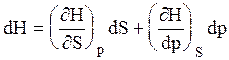 ,
,
а также используя выражение (16)
 (6.129)
(6.129)
Аналогично для внутренней энергии U = f (S,V)
 .
.
С другой стороны dU = TdS – pdV.
 (6.130)
(6.130)
Полученные выражения (6.127)-(6.130) показывают сущность метода характеристических функций, т.е. частные производные от функции состояния системы при соответствующем выборе независимых переменных дают в простом и явном виде термодинамические параметры системы.
Соотношения (6.127-6.130) имеют большое значение при решении различных физико-химических задач.
Для запоминания характеристических функций можно воспользова-ться диаграммами, так называемыми мнемоническим кругом и мнемоническим квадратом, представленными на рис. 6.17.
 V T V
V T V
Рис. 6.17. Мнемонический квадрат и мнемонический круг для определения
частных производных характеристических функций
Характеристические функции F (T,V), G (T,p), H (S,p) и U (S,V) обладают следующими свойствами:
а) все они являются функциями состояния, т.е. изменение этих функций при переходе системы из одного состояния в другое не зависит от пути перехода, а определяется значениями этих функций в начальном и конечном состоянии, т.е.
б) функции F, G, H, U являются аддитивными, т.е. значения их зависят от размеров системы, пропорциональны числу моль или массе;
в) все они имеют размерность энергии, т.е. измеряются в Дж, Дж/моль, кДж;
г) физический смысл изменения характеристических функций — есть работа некоторого процесса при определенных условиях, согласно выражениям (6.114), (6.116), (6.118) и (6.119);
д) при переходе системы в состояние равновесия характеристические функции принимают экстремальное значение;
е) характеристические функции взаимосвязаны.
Последнее свойство имеет большое значение для физико-химических исследований.
Взаимосвязь функций U, F, G, H хорошо видна на диаграмме соотношений между ними (рис. 6.18)
 H
H
Рис. 6.18. Соотношение между характеристическими функциями
На рис. 6.18 характеристические функции изображены в виде отрезков, длины которых находятся из следующих соотношений
H = U + pV = G + TS = F + pV + TS
F = U – TS = G – pV (6.131)
G = H – TS = U + pV – TS
Используя соотношения (6.131) и значения частных производных (6.127), (6.128) можно получить уравнения Гиббса-Гельмгольца, которые также отражают взаимосвязь характеристических функций.
Получим уравнения Гиббса-Гельмгольца.
По определению G = H – TS, (6.113), но из (6.117) имеем

Подставляя (6.127) в (6.113) получаем  ,
,
т.е. связь таких термодинамических функций, как энергия Гиббса и энтальпия системы.
Если рассматривать изменение этих функций в каком-либо процессе, то получим первое уравнение Гиббса-Гельмгольца, а именно
 (6.132)
(6.132)
Соотношение (6.132) устанавливает связь между тепловым эффектом химической реакции, протекающей при постоянном давлении и макси-мальной полезной работе изобарно-изотермического процесса, т.е.
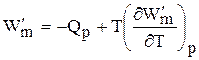 , (6.133)
, (6.133)
Аналогично для свободной энергии Гельмгольца, согласно (6.115)
 (согласно 6.118).
(согласно 6.118).
Подставляя (6.128) в (6.115), получаем:  ,
,
т.е. связь свободной энергии Гельмгольца (F) и внутренней энергии системы (U), а также
 (6.134)
(6.134)
Выражение (6.134) устанавливает связь между тепловым эффектом химической реакции, протекающей при постоянном объеме, и максимальной полезной работой изохорно-изотермического процесса, т.е.

 , (6.135)
, (6.135)
т.к. DU = QV, а –DF = W¢m (согласно 40)
Теплоты Qp и QV в выражениях (6.133) и (6.135) относятся не к процессам, которым соответствует работа W¢m.
W¢m — это работа обратимого процесса, а Qp = DH и Q = DU- тепловые эффекты химических реакций или физико-химических процессов, когда они совершаются предельно необратимо, но между теми же начальным и конечным состояниями системы.
Теплоты же равновесного процесса равные Qобр = TDS, выражаются последними членами уравнений 6.132, 6.133, 6.134, 6.135.
Исходя из изложенного, можно записать
Уравнения 6.133 и 6.134 можно привести к виду, удобному для интегрирования.
Для этого разделим обе части уравнений на Т 2 и выполним преобразования:


Тогда получаем 
или 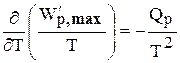 ,
, 
Интегрирование уравнений (6.136) дает следующие выражения:

Соотношения (6.137) позволяют вычислить максимальную полезную работу изобарического (W¢p,max) и изохорического (W¢V,max) процессов, зная его теплоту Qp или QV, если возможно найти постоянную интегрирования.
Уравнения Гиббса-Гельмгольца широко используются в физико-химических расчетах, например, для вычисления тепловых эффектов химических реакций по опытным значениям электродвижущих сил электрохимических элементов, для вычисления DS и других термодинами-ческих величин.
Основные понятия химического равновесия и процессы прогнозирования свойств получаемых материалов
Общие соотношения, вытекающие из второго закона термодина-мики, позволяют определить условия химического равновесия. Более простым случаем является равновесие при химических реакциях, протекающих без фазовых превращений, т.е. реакциях, когда исходные вещества и продукты находятся в одной фазе. Например, реакции между газами или реакции в растворах.
Химические процессы, играющие важную роль при создании материалов, как правило, не протекают до полного исчезновения исходных веществ, и останавливаются, достигая определенного состояния химии-ческого равновесия.
Основной целью синтеза любого материала является максимальное достижение выхода конечных продуктов, поэтому важно уметь характе-ризовать равновесие и найти параметры, от которых оно зависит, чтобы управлять процессом в целом.
Одним из важных вопросов химической термодинамики является вопрос о возможности и направленности протекания самопроизвольного процесса и условии равновесия.
Возможность, направление и предел самопроизвольного протекания процессов перехода энергии или вещества от одной части системы к другой зависят только от соотношения факторов интенсивности. Такими факторами являются температура (критерий перехода теплоты), давление (критерий перехода газа), потенциал заряда (критерий перехода электричества) и т.д.
Критерием самопроизвольного перехода данного компонента из одной фазы в другую является химический потенциал данного компонента, который тоже относится к факторам интенсивности. При умножении его на изменение количества этого компонента дает изменение соответствующего вида энергии. Согласно II закону термодинамики самопроизвольное протекание процессов взаимодействия между разными частями системы возможно только в направлении выравнивания фактора интенсивности (температуры, давления, химического потенциала и т.д.) для всех частей системы.
Достижение одинакового значения этого фактора является пределом самопроизвольного течения процессов в данных условиях и, следовательно, условием равновесия. Так, условием равновесия между фазами a и b компонента i служит равенство химического потенциала этого компонента в обеих фазах.
Этот вывод относится к любому компоненту, входящему в состав фаз, и к любым фазам системы.
Таким образом, из какого бы числа компонентов и из какого бы числа фаз ни состояла гетерогенная система условием равновесия между фазами в ней является то, что химический потенциал любого данного компонента должен быть одинаковым во всех фазах системы.
Химический потенциал
До сих пор мы рассматривали системы, сохраняющие постоянной массу, в которых изменение энергии было следствием сообщения или отнятия теплоты или результатом совершения работы.
Течение многих процессов сопровождаются изменением числа моль компонентов в системе. Так, например, любая гомогенная химическая реакция протекает с уменьшением числа моль исходных веществ и увеличением продуктов реакции, при фазовых переходах один компонент переходит из одной фазы в другую, в результате количество его уменьшается в одной фазе и увеличивается в другой. С помощью эксперимента установлено, что любое химическое изменение системы зависит от природы реагентов, состава исходных веществ и продуктов реакции, от концентрации реагентов и от внешних условий (Т, Р).
А любое изменение химического состава системы с термодинамической точки зрения является результатом перераспределения масс отдельных компонентов в системе. Термодинамическое описание таких систем, в которых происходит изменение массы взятых веществ, имеет большое практическое значение.
Изменение количества вещества в системе (появление или удаление его) приводит к изменению энергии системы.
Допустим, что увеличивается масса одного лишь компонента на dn1, тогда внутренняя энергия (U) увеличивается на величину, пропорциональную dn1. Коэффициент пропорциональности обозначим через m (его размерность  ). В этом случае основное уравнение термодинамики
). В этом случае основное уравнение термодинамики
При изменении числа моль компонентов в системе внутренняя энергия будет меняться на величину mdn.
Основное уравнение термодинамики с переменным составом будет выглядеть так:
или DU = TDS – pDV + mDn (6.140)
Решив его относительно коэффициента m получим важное соотношение:
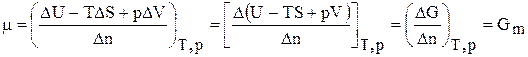 , (6.141)
, (6.141)
где Gm — мольное значение функции G для данного вещества, которое для чистого вещества является его химическим потенциалом.
Химический потенциал вещества зависит от межмолекулярных сил в системе, а в индивидуальном веществе проявляются силы взаимодействия между одинаковыми молекулами А-А. Если система представляет смесь веществ А и В, то возможны и другие взаимодействия между молекулами А-В и В-В.
Если система, в которой происходит изменение числа моль компонентов, не совершает никаких других видов работы, кроме работы расширения, то из сравнения уравнения (6.139) и уравнения:
dU = TdS – pdV – dW¢max
Следовательно, величина midni имеет смысл химической работы, которая совершается при протекании химической реакции.
Любой вид работы можно представить в виде произведения фактора интенсивности (силы, давления, поверхностного натяжения и т.д.), не зависящего от массы, на изменение фактора экстенсивности (емкости):
dWэлектрич. = – EdP (P — поляризация)
dWобраз. единицы поверхности = – sdS; (где S — поверхность)
Таким образом, dn — изменение фактора экстенсивности, а m — фактор интенсивности. Во всех случаях взаимодействия систем с различными потенциалами происходит их выравнивание за счет изменения соответствующих факторов емкостей. Например, давление выравнивается за счет изменения объема и т. д. Выравнивание химических потенциалов происходит за счет изменения масс веществ. Но в отличие от Р и Т, которые могут быть измерены, химический потенциал не может быть измерен непосредственно. Тем не менее, химический потенциал — важная термодинамическая функция, которую применяют для изучения равновесия в различных термодинамических системах. Понятие о химическом потенциале введено в химию Дж. Гиббсом в 1878 году.
Если рассматривать смеси переменного состава и записать уравнения других термодинамических потенциалов:
то из этих уравнений также следует, что химический потенциал можно выразить через любую характеристическую функцию:

 ,
,
где ni — постоянное количество всех компонентов;
nj — постоянное количество всех компонентов, кроме рассматриваемого; i ¹ j .
Т.е. химический потенциал данного вещества равен частной производной от любого термодинамического потенциала по числу моль данного вещества при соответствующих условиях.
Чаще всего изучаются системы при p, T = const, поэтому в дальнейшем химический потенциал i-того компонента будем определять как приращение энергии Гиббса при добавлении одного моль этого компонента к большому объему системы при постоянных температуре и давлении.

Понятие «большой объем системы» означает, что состав системы практически не изменяется после добавления одного моль компонента. Как уже указывалось, химический потенциал чистого вещества равен энергии Гиббса одного моль этого вещества: mi = Gi, т.к. при изменении количества чистого вещества на один моль, энергия Гиббса изменится на величину, равную энергии Гиббса одного моль вещества. Если в системе содержится “n” моль компонента, то
Итак, во всех практически важных расчетах вычисление химическо-го потенциала связано с определением функции G для изучаемой системы.
Рассмотрим условия равновесия и самопроизвольности процессов, связанных с изменением состава. Запишем основное уравнение термо-динамики таких систем, когда в системе содержится k компонентов
Т.к. в изобарно-изотермической системе условием самопроизвольности является dG
http://poisk-ru.ru/s43997t1.html
http://lektsia.com/5×3883.html