Решение уравнения Шредингера для многоэлектронных атомов
Автор работы: Пользователь скрыл имя, 13 Октября 2013 в 00:54, реферат
Краткое описание
Уравнение Шрёдингера для атомов, содержащих более одного электрона, не может быть решено в аналитическом виде. В связи с этим рассматривают приближённые методы, наиболее существенным, из которых является метод самосогласованного поля. Идея метода заключается в том, что каждый электрон в атоме рассматривается как движущийся в самосогласованном поле, создаваемом ядром вместе со всеми остальными электронами.
Содержание
Введение…………………………………………………………………………….3
Многоэлектронные атомы…………………………………………………………4
Методы решения уравнения Шредингера для многоэлектронных атомов
1.1 . Метод самосогласованного поля Хартри…………………………………. 5
1.2. Принцип Паули и определители Слейтора. Метод Хартри – Фока………7
Вывод………………………………………………………………………………10
Литература………………………………………………………………………. 11
Прикрепленные файлы: 1 файл
сдать2.doc
ГОСУДАРСТВЕННОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
«ВОРОНЕЖСКИЙ ГОСУДАРСТВЕННЫЙ ПЕДАГОГИЧЕСКИЙ УНИВЕРСИТЕТ»
РЕШЕНИЕ УРАВНЕНИЯ ШРЕДЕНГЕРА ДЛЯ МНОГОЭЛЕКТРОННЫХ АТОМОВ
Выполнила студентка 4 курса 2 группы естественно-географического
факультета отделения «Химия-биология»
Проверила доцент кафедры химии
Методы решения уравнения Шредингера для многоэлектронных атомов
1.2. Принцип Паули и определители Слейтора. Метод Хартри – Фока………7
Введение.
Уравнение Шрёдингера для атомов, содержащих более одного электрона, не может быть решено в аналитическом виде. В связи с этим рассматривают приближённые методы, наиболее существенным, из которых является метод самосогласованного поля. Идея метода заключается в том, что каждый электрон в атоме рассматривается как движущийся в самосогласованном поле, создаваемом ядром вместе со всеми остальными электронами. Вместе с тем этот метод может применяться не только в атомной физике, но и просто для систем взаимодействующих частиц.
Построение самосогласованного поля может осуществляться либо методом последовательных приближений (изначально предложенным Хартри) или прямым вариационным методом.
Существенно, что вычисления методом самосогласованного поля весьма громоздки, особенно для сложных атомов. Для них применяется другой метод — метод Томаса — Ферми.
Обобщение метода Хартри — Фока, в котором учитываются волновые функции пар частиц, является метод Хартри — Фока — Боголюбова.
В атоме водорода электрон находится в силовом поле, которое создается только ядром. В многоэлектронных атомах на каждый электрон действует не только ядро, но и все остальные электроны. При этом электронные облака отдельных электронов как бы сливаются в одно общее многоэлектронное облако. Точное решение уравнения Шредингера для таких сложных систем связано с большими затруднениями и, как правило, недостижимо. Поэтому состояние электронов в сложных атомах и в молекулах определяют путем приближенного решения уравнения Шредингера.
Общим для всех приближенных методов решения этого уравнения является так называемое одноэлектронное приближение, т. е. предположение, что волновая функция многоэлектронной системы может быть представлена в виде суммы волновых функций отдельных электронов. Тогда уравнение Шредингера может решаться отдельно для каждого находящегося в атоме электрона, состояние которого, как и в атоме водорода, будет определяться значениями квантовых чисел n, l, m и s. Однако и при этом упрощении решение уравнения Шредингера для многоэлектронных атомов и молекул представляет весьма сложную задачу и требует большого объема трудоемких вычислений. В последние годы подобные вычисления выполняются, как правило, с помощью быстродействующих электронных вычислительных машин, что позволило произвести необходимые расчеты для атомов всех элементов и для многих молекул.
Исследование спектров многоэлектронных атомов показало, что здесь энергетическое состояние электронов зависит не только от главного квантового числа n, но и от орбитального квантового числа l. Это связано с тем, что электрон в атоме не только притягивается ядром, но и испытывает отталкивание со стороны электронов, расположенных между данным электроном и ядром. Внутренние электронные слои как бы образуют своеобразный экран, ослабляющий притяжение электрона к ядру, или, как принято говорить, экранируют внешний электрон от ядерного заряда. При этом для электронов, различающихся значением орбитального квантового числа l, экранирование оказывается неодинаковым.
В многоэлектронных атомах энергия электрона зависит не только от главного, но и от орбитального квантового числа. Главное квантовое число определяет здесь лишь некоторую энергетическую зону, в пределах которой точное значение энергии электрона определяется величиной l. В результате возрастание энергии по энергетическим подуровням происходит примерно в следующем порядке:
ls 2 \rij в уравнении , зависящей от координат двух электронов, выражающим, описывающим межэлектронного взаимодействия как функцию координат каждого отдельного электрона: Vэф.
Чтобы определить особые функции ѱi и особые значения оператора гамильтона надо знать потенциал Vэф.( ѱi), который зависит от искомых функций. Эта трудность устраняется в методе последовательных приближений. В качестве начальных волновых функций берут какие-либо пробные функции, например, функции водородоподобного атома.
Затем решают уравнение Шредингера и находят функции первого приближения усредненного потенциала. Обычно новые величины энергий межэлектронного взаимодействия сильно отличается от первоначальных, что связано с неточностью исходных функций. Поэтому находят функции следующего приближения и т.д.
Критерием получения достаточно хороших ѱi является совпадение с заданной точностью величин Vэф., рассчитанных для ѱ n и ѱ (n+1) т.е. потенциалы Vэф. должны быть согласованными с функциями ѱi . Это требование и определяет название метода самосогласованного поля.
Уравнение Шредингера
Вы будете перенаправлены на Автор24
Предпосылки вывода уравнения Шредингера
Основная идея волновой механики заключается в том, что для таких малых тел, как электрон, нельзя с определенностью сказать, где оно находится в данное время и куда направляется. Можно установить только относительную вероятность его нахождения в том или ином месте и наличие определенного количества движения в определенный момент времени.
В соответствии с волновой механикой какая-либо система – атом, молекула, электрон и т.д. – описывается функцией состояния или волновой функцией, обозначаемой $\psi$ («пси»), которая является функцией координат всех частиц, образующих эту систему. Следовательно, величина $\psi$ зависит только от положения всех частиц в пространстве.
В 1924 г. де Бройль предположил, что точно также, как свет, который, как обычно считают, имеет волновую природу, на самом деле при определенных обстоятельствах ведет себя, как будто он состоит из частиц – квантов, — так и очень малые частицы, такие, как электроны, также могут обладать волновыми свойствами. Де Бройль предположил, что с пучком электронов следует связывать длину волны, определяемую уравнением
где $\hbar$ – постоянная Планка ($6,626\cdot 1034 Дж\cdot с$ или $6,626\cdot 10-27 эрг\cdot с$), а $p$ – количество движения (импульс) электрона в пучке, т.е. его масса, умноженная на его скорость.
Физическое подтверждение волновой природы электрона было продемонстрировано в 1927 – 1928 гг. Дейвиссоном, Джермером и Томсоном, которые показали, что пучок электронов может испытывать дифракцию на подходящей решетке (атомы в кристалле золота), аналогичную дифракции пучка света.
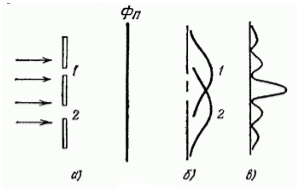
Рисунок 1. Дифракция пучка электронов
На преграду с двумя узкими щелями направлен параллельный пучок моноэнергетических (т.е. обладающих одинаковой кинетической энергией) электронов (рис. 1. а). За преградой находится фотопластина $Фn$. При закрытии щели номер $2$ и экспонировании в течение времени $t$ почернение на проявленной фотопластине будет характеризоваться кривой $1$ (рис. 1. б). При закрытии щели номер $1$, соответственно, почернение на фотопластине будет соответствовать кривой $2$. Однако в случае, когда открыты обе щели картина почернения фотопластины (рис. 1. в) отнюдь не эквивалентна наложению двух первых картин. Зато она аналогична картине, получающейся при интерференции двух когерентных световых волн.
Готовые работы на аналогичную тему
Тот факт, что системы малых частиц проявляют, по крайней мере, при определенных условиях, волновые свойства, предполагает возможность описания таких систем уравнениями, подобными те, которые описывают другие виды волнового движения, например, волны, которые распространяются вдоль колеблющейся струны, или волновое движение, приписываемое электромагнитному излучению. Действительно, можно начать с волнового уравнения, соответствующего электромагнитным волнам, и путем определенных замен, превратить его в уравнение, соответствующее нашему случаю. Хотя эти замены диктуются физическими причинами, они в основном произвольны и могут быть приняты только потому, что приводят к уравнению, которое, как показывает опыт, позволяет получить правильное решение физических задач. Поэтому следует принять волновое уравнение как постулат, так как у химиков основной интерес вызывает применение волнового уравнения к атомным и молекулярным системам, а не физические и математические соображения, которыми руководствовался Шредингер, впервые его предложивший в 1925 г.
Общий вид уравнения Шредингера

Рисунок 2. Эрвин Шрёдингер (1887 — 1961)
Волновое уравнение, применяемое для расчета стационарных состояний системы, можно записать в символическом виде:
где $H$ представляет собой определенный способ выражения общей энергии системы, а $E$ – числовое значение этой энергии. Для всех систем, которые обычно интересуют химиков, общая энергия представляет собой сумму кинетической энергии $Т$ и потенциальной энергии $V$:
Это соотношение было широко использовано физиком-теоретиком Гамильтоном, поэтому $H$ часто называют функцией Гамильтона, а $\mathcal H$ гамильтонианом системы.
Уравнение Шредингера на примере атома водорода
Рассмотрим модель атома водорода, предложенную Бором. Для простоты предположим, что тяжелое ядро закреплено (оно почти, но не совершенно неподвижно, когда электрон движется вокруг него). Тогда полная кинетическая энергия $Т$ системы представляет собой просто кинетическую энергию электрона
где $m$ – масса электрона и $\nu$ – его скорость. Потенциальная энергия системы есть просто энергия, возникающая вследствие электростатического взаимодействия (гравитационные силы приблизительно в $10^<18>$ раз меньше), и ее можно выразить как
где $e$ — заряд электрона, $r$ — радиус орбиты, знак минус появляется вследствие того, что заряд одной из частиц положителен $(+)$, а другой отрицателен $(-)$. Поэтому для атома водорода функция Гамильтона в классической (т.е. доквантовомеханической) физике равна:
Если использовать понятие количества движения электрона $p=m\nu$, данное уравнение запишется в следующем виде:
Теперь для перехода от классического описания этой или какой-либо другой системы к описанию при помощи волновой механики, необходимо взять функцию Гамильтона (уравнение 6) и произвести в ней определенные замены: в функции Гамильтона количество движения следует заменить выражением
Таким образом, гамильтониан для атома водорода в его квантовомеханической форме $<\mathcal H>$ следует записать в виде
Если теперь это выражение гамильтониана подставить в общее волновое уравнение (уравнение 1), то получим:
Это и есть волновое уравнение для атома водорода. Из уравнения 9 следует, что нужно вторые производные функции $\psi $ сложить и умножить на $-<<\hbar >^2>/<8<\pi >^2m>$, затем к этому добавить $\left(-
Лекция №1. Квантов0механическ0е обоснование теории строения молекул и химической связи. Строение атома (стр. 4 )
 | Из за большого объема этот материал размещен на нескольких страницах: 1 2 3 4 5 6 7 8 9 |

Из различных методов определения СЭ наиболее прямой и точный — измерение минимальной энергии фотоотрыва электрона от отрицательного иона.
Для большинства атомов присоединение электрона — экзотермический процесс. Наиболее высоким по абсолютной величине сродством к электрону обладают атомы галогенов в последовательности Cl > F > Вг > I. Энергии ионизации и сродство к электрону молекул определяют также, как это сделано для атома.
ТЕОРЕТИЧЕСКИЕ МЕТОДЫ, ПРИМЕНЯЕМЫЕ ПРИ ИЗУЧЕНИИ СТРОЕНИЯ МОЛЕКУЛ И ХИМИЧЕСКОЙ СВЯЗИ
1. Молекула. Потенциальная поверхность. Равновесная конфигурация
2. Теория химической связи и её задачи.
3. Вариационный метод решения уравнения Шрёдингера.
§1. Молекула. Потенциальная поверхность. Равновесная конфгурация.
В химии молекулой называют наименьшую частицу данного вещества, обладающую его химическими свойствами, способную к самостоятельному существованию. Если отвлечься от поступательного движения молекулы как целого, то в ее энергию вносят вклад три вида движения: 1) движение электронов в поле ядер, 2) колебание ядер около положения равновесия и вращение молекулы вокруг оси, проходящей через центр масс, причем Еэл >> Екол >>Евр.
Хотя эти движения взаимосвязаны, можно приближенно рассматривать их как независимые и считать энергию молекулы равной сумме электронной, колебательной и вращательной энергий:
Е = Еэл + Екол +Евр. (6.1)
Волновая функция молекулы в этом приближении равна произведению функций, описывающих указанные три вида движения:
 (6.2)
(6.2)
Остановимся на наиболее важной составляющей энергии молекулы — электронной энергии. Так как скорость тяжелых ядер во много раз меньше скорости легких электронов, приближенно можно рассматривать движение электронов в молекуле в каждый данный момент, считая ядра неподвижными (приближение Борна — Оппенгеймера). Выбранному фиксированному положению ядер R отвечает определенная энергия электронов  , включающая их кинетическую энергию, энергию взаимодействия электронов друг с другом и энергию взаимодействия электронов с ядрами.
, включающая их кинетическую энергию, энергию взаимодействия электронов друг с другом и энергию взаимодействия электронов с ядрами.
 Условимся включать сюда также энергию отталкивания ядер
Условимся включать сюда также энергию отталкивания ядер  . Тогда название «электронная» для
. Тогда название «электронная» для  указывает, что учитывается движение только электронов, но не ядер, а фиксированное расстояние между ядрами R рассматривается как параметр. Индекс «эл» при этом можно опустить (Eкол и Евр здесь не рассматриваются). Если расстояние между ядрами R изменится, изменится поле ядер, в котором движутся электроны, изменится и электронная энергия системы
указывает, что учитывается движение только электронов, но не ядер, а фиксированное расстояние между ядрами R рассматривается как параметр. Индекс «эл» при этом можно опустить (Eкол и Евр здесь не рассматриваются). Если расстояние между ядрами R изменится, изменится поле ядер, в котором движутся электроны, изменится и электронная энергия системы  . В этом смысле электронная энергия есть функция межъядерного расстояния и по отношению к движению ядер играет роль потенциальной энергии. Вид функции для двухатомной молекулы АВ изображает кривая о рис. 5.1., называемая потенциальной кривой. Когда атомы А и В удалены на бесконечное расстояние, электронная энергия
. В этом смысле электронная энергия есть функция межъядерного расстояния и по отношению к движению ядер играет роль потенциальной энергии. Вид функции для двухатомной молекулы АВ изображает кривая о рис. 5.1., называемая потенциальной кривой. Когда атомы А и В удалены на бесконечное расстояние, электронная энергия  равна сумме электронных энергий невзаимодействующих атомов А и В в основном состоянии:
равна сумме электронных энергий невзаимодействующих атомов А и В в основном состоянии:  .
.
При сближении ядер электронная энергия понижается (сила притяжения электронов к ядрам преобладает над силами отталкивания между ядрами и между электронами). Затем потенциальная кривая проходит через минимум при R = rе и при дальнейшем сближении ядер возрастает, стремясь к бесконечности при R →0 (преобладает сила отталкивания). Межъядерное расстояние R=re, отвечающее минимуму потенциальной кривой, называется равновесным. При R=re равнодействующая всех сил притяжения и отталкивания равна нулю, молекула находится в устойчивом состоянии. Этому состоянию отвечает строго определенное значение электронной энергии молекулы. За нуль отсчета энергии можно принять, как и для атома, энергию невзаимодействующих электронов и ядер. Разность между суммой энергий невзаимодействующих атомов и Eэл. мол называют энергией химической связи или энергией диссоциации молекулы, отсчитанной от минимума потенциальной кривой, и обозначают De (глубина «потенциальной ямы»):
Данная потенциальная кривая соответствует классическим представлениям. Однако в нее надо внести поправки, так как равновесное состояние неосуществимо с точки зрения квантовой механики: в этом состоянии ядра неподвижны, значит, одновременно точно определены координата (R = rе) и импульс (р = 0), что противоречит соотношению неопределенностей Гейзенберга. Параметры rе и De (рис. 6.1) относятся таким образом к гипотетическому равновесному состоянию. В действительности даже и при 0К ядра не зафиксированы при R — rе, а совершают колебания около положения равновесия. Реальная энергия молекулы при этом выше, чем предполагалась, на величину энергии «нулевых колебаний»  (рис.6.1). При нулевых колебаниях расстояние R между ядрами изменяется, в результате чего нулевой колебательный уровень характеризуется некоторым усредненным значением r0, которое из-за асимметрии потенциальной кривой незначительно, отличается от гипотетического равновесного расстояния rе=0,751·10-10 м и 0,741·10-10 м соответственно для Н2).
(рис.6.1). При нулевых колебаниях расстояние R между ядрами изменяется, в результате чего нулевой колебательный уровень характеризуется некоторым усредненным значением r0, которое из-за асимметрии потенциальной кривой незначительно, отличается от гипотетического равновесного расстояния rе=0,751·10-10 м и 0,741·10-10 м соответственно для Н2).
Определяемая на опыте энергия диссоциации молекулы D0 отсчитывается не от минимума потенциальной кривой как De, а от уровня нулевых колебаний (рис.и связана с De соотношением
 (6.4)
(6.4)
Энергия диссоциации D0, служит мерой прочности химической связи и определяется как изменение энергии в процессе — АВ = А + В при 0 К в идеально-газовом состоянии. Если специальных указаний нет, то понимается, что как молекула АВ, так и атомы А и В находятся в основном электронном состоянии. Это определение сохраняет силу и для многоатомных молекул. Например, для молекулы AmBn энергией диссоциации будет изменение энергии в процессе AmBn = mA + nB.
Потенциальная поверхность. Равновесная конфигурация. При описании потенциальной кривой вместо Eэл. мол обычно используют символ E(R) или Е. Для многоатомной молекулы Е является функцией уже не одной, а нескольких пространственных координат Rij. Например, потенциальная энергия молекулы ABC является функцией трех независимых координат — R1(A — В), R2(B — С) и угла о(АВС) или расстояний R1(A — В), R2(B — С) и R3(А — С).
Для линейной молекулы с фиксированным углом α- 180° эта функция изобразится поверхностью в трехмерном пространстве (потенциальная поверхность) Устойчивому состоянию молекулы отвечает минимальное значение ее энергии E(АВС) и определенное относительное расположение ядер в пространстве, называемое равновесной конфигурацией молекулы с параметрами rе(А — В) и rе(В — С). Глубина потенциальной ямы определяет энергию химической связи De и по формуле (5.4) энергию диссоциации молекулы D0.
Для более сложной молекулы, чем линейная ABC, равновесная конфигурация и энергия равновесного состояния определяются положением минимума на потенциальной поверхности в многомерном пространстве. Если потенциальная поверхность имеет два (или более) минимума, для молекулы возможны два изомера или более, отличающиеся параметрами равновесной конфигурации и энергией. Если минимума на потенциальной поверхности нет, данная система нестабильна, при любом расположении ядер она распадается на невзаимодействующие атомы.
Так же как и атом, молекулу можно перевести в возбужденные электронные состояния (энергия возбуждения Те), каждому из которых отвечает своя потенциальная поверхность или кривая (кривая б на рис. 6.1).
Рассмотрев потенциальную кривую (поверхность), можно дать еще одно определение молекулы молекула — физически устойчивая система, состоящая из двух (или более) ядер и определенного числа электронов, состояние которой описывается потенциальной кривой (поверхностью) с минимумом.
Говоря о физической устойчивости, понимают, что соединение атомов в молекулу сопровождается понижением энергии системы. Данным здесь определением охватываются кроме обычных молекул (Н2, СН4 и др.) также радикалы (СН, ОН, СН3 и др.) и молекулярные ионы (Н+, O2— и др.). Этому отвечает одинаковый подход теории строения к изучению перечисленных типов частиц. В тех случаях, когда молекулы одноатомны (благородные газы, пары металлов), сохраняет силу аналогичное определение для атома.
Равновесная конфигурация предполагает жесткую фиксацию всех межъядерных расстояний в молекуле. Однако реальная молекула не является жесткой системой. Вместе с тем у огромного большинства молекул амплитуды колебаний ядер весьма малы по сравнению с межъядерными расстояниями и можно, пренебрегая колебаниями, рассматривать молекулы как жесткие системы («квазижесткие» или «квазитвердые» молекулы).
Равновесные конфигурации молекул принято относить к тем или иным точечным группам симметрии.
Двухатомные молекулы подразделяются на молекулы с одинаковыми ядрами, или гомонуклеарные (например, Н2), и с неодинаковыми ядрами, или гетеронуклеарные (например, HCI). Свойства симметрии их различны.
Симметрия равновесной конфигурации определяет и симметрию электронного облака молекулы. В связи с этим гомонуклеарные и гетеронуклеарные молекулы различаются по электрическим и оптическим свойствам, таким, как дипольный момент, поляризуемость и магнитная восприимчивость, правила отбора в спектрах. То же относится и к многоатомным молекулам, различающимся по симметрии, как, например, СН4 и СН3С1.
Рассмотренные молекулярные параметры: энергия диссоциации, межъядерные расстояния, равновесная конфигурация важны для химии не только как индивидуальные характеристики молекул. По ним можно рассчитать термодинамические свойства веществ и константы равновесия химических реакций.
§2. Теория химической связи и ее задачи. Уравнение Шредингера для молекул
Взаимодействие атомов, приводящее к образованию молекул простых и сложных веществ, а также кристаллов, называют химической связью. Взаимодействие атомов многообразно, поэтому многообразны и химические связи, которые часто сводят к нескольким основным типам: ковалентной, ионной, донорно-акцепторной, водородной связи и др. Однако все эти взаимодействия можно описать с позиций единой теории химической связи.
1) Эта теория призвана объяснить, какие силы действуют между атомами, как атомы объединяются в молекулы, что обеспечивает устойчивость образовавшейся сложной частицы (то же относится к кристаллам, жидкостям и другим телам).
2) Теория должна объяснить опытные факты, лежащие в основе классического понятия валентности, и наряду с этим существование и устойчивость многочисленных соединений, не укладывающихся в привычные рамки классических структурно-химических представлений.
3) Теория должна разработать единые методы расчета молекулярных параметров, интерпретировать молекулярные спектры.
4) Наконец, теория должна сделать возможным априорный расчет скорости химического процесса, зависимости ее от строения молекул реагирующих веществ.
 |
Современная теория химической связи, теория строения молекул и кристаллов базируется на квантовой механике: молекулы, как и атомы, построены из ядер и электронов, и теория химической связи должна учитывать корпускулярно-волновой дуализм микрочастиц. До применения методов квантовой механики к химии не удавалось создать непротиворечивую теорию химической связи.
Её фундамент был заложен в 1927 г. Гейтлером и Лондоном. Выполнив на основе квантовой механики расчет свойств молекулы водорода, они показали, что природа химической связи электрическая, никаких особых сил химического взаимодействия не существует. Действующие в молекуле между ядрами и электронами гравитационные и магнитные силы пренебрежимо малы по сравнению с электрическими силами.
Квантовомеханический подход к исследованию строения атома и молекулы один и тот же: нужно составить и решить уравнение Шредингера для системы из электронов и ядер и дать физическую интерпретацию полученным решениям. Составляя уравнение Шрёдингера для электронной энергии молекулы в приближении Борна — Оппенгеймера, считают ядра неподвижными.
Следовательно, электронная энергия для молекулы не зависит от координат ядер, а только от фиксированного расстояния R между ними (рис. 6.2). Во внимание принимаются лишь изменения координат электронов. Простейшая из молекул молекулярный ион  содержит один электрон и два ядра. Для одного электрона в поле двух ядер (ион ) уравнение Шредингера имеет вид
содержит один электрон и два ядра. Для одного электрона в поле двух ядер (ион ) уравнение Шредингера имеет вид
 . (6.5)
. (6.5)
Для молекулярного иона потенциальная энергия
 (6.6)
(6.6)
включает притяжение электрона 1 к ядрам А и В (первые два члена) и отталкивание ядер (ядра А и В у совершенно одинаковы — это протоны). Для иона  с учетом (6.6) уравнение Шредингера принимает вид
с учетом (6.6) уравнение Шредингера принимает вид
 , (6.7)
, (6.7)
где Ψ — одноэлектронная волновая функция (собственная функция Шредингера для системы с одним электроном). Индексы «эл. мол.» при E и Ψ — опущены для упрощения записи.
Состояние молекулы Н2 описывается уже двухэлектронной функцией Ψ, зависящей от координат двух электронов (см. рис. 6.2).
Многоэлектронными будут и волновые функции более сложных молекул. Уравнение Шредингера для молекулы Н2 имеет вид
 (6.8)
(6.8)
Индексы 1 и 2 при операторах Лапласа указывают, что волновая функция молекулы  дифференцируется по координатам первого (
дифференцируется по координатам первого ( ) и второго (
) и второго ( ) электронов. Усложнится и выражение для потенциальной энергии:
) электронов. Усложнится и выражение для потенциальной энергии:
 (6.9)
(6.9)
В (6.9) первые четыре члена обозначают потенциальную энергию притяжения электронов 1 и 2 к ядрам А и В соответственно, пятый член — потенциальную энергию взаимного отталкивания электронов 1 и 2, последний член — энергию отталкивания ядер. Аналогично записывается уравнение Шредингера и для многоатомных молекул. В уравнениях (6.7) и (6.8) используется координатная молекулярная функция Ψ. Полная волновая функция молекулы Фмол, учитывающая и спин, должна удовлетворять принципу Паули антисимметрии волновых функций и строится в виде определителя.
Для молекулы, так же как и для атома, точное решение уравнения Шредингера возможно лишь для системы, содержащей один электрон — для молекулярного иона типа  . Уже для молекулы Н2 в выражении (6.9) появляется член
. Уже для молекулы Н2 в выражении (6.9) появляется член  (энергия электронного отталкивания), зависящий от координат двух электронов, и точное решение невозможно. Однако принципиальный ответ о природе химической связи можно получить и при помощи приближенных методов решения волнового уравнения.
(энергия электронного отталкивания), зависящий от координат двух электронов, и точное решение невозможно. Однако принципиальный ответ о природе химической связи можно получить и при помощи приближенных методов решения волнового уравнения.
§3. Вариационный метод решения уравнения Шрёдингера
Одним из широко применяемых при рассмотрении теории химической связи приближенных методов решения уравнения Шредингера является вариационный метод. Здесь коротко излагается его сущность.
Уравнение Шредингера (1.1) может быть представлено в так называемой операторной форме. Для этого разделим все члены уравнения на  и перегруппируем члены. Получим
и перегруппируем члены. Получим
 .
.
Сумма двух действий, производимых над функцией Ψ в левой части, дифференцирование и умножение, может быть записана с помощью оператора  ,
,

который называют оператором энергии, оператором Гамильтона или гамильтонианом. Уравнение приобретает лаконичную форму:
По такому же принципу строится оператор Гамильтона для многоатомных систем, например, для молекулы Н2
 (6.11)
(6.11)
Умножим обе части уравнения Шредингера (6.10) на функцию Ψ*, комплексно сопряженную с волновой функцией
 . (6.12)
. (6.12)
Если Ψ — действительная функция, то Ψ*=-Ψ. (Левую сторону уравнения (6.12) нельзя записать аналогично правой в виде  , так как в отличие от E
, так как в отличие от E  — оператор, а не число). Возьмем интеграл от обеих частей (5.12) по всему пространству, причем Е как постоянную величину вынесем за знак интеграла:
— оператор, а не число). Возьмем интеграл от обеих частей (5.12) по всему пространству, причем Е как постоянную величину вынесем за знак интеграла:
 .
.
 . (6.13)
. (6.13)
Если функция Ψ нормирована, знаменатель обращается в единицу и
 . (6.14)
. (6.14)
Если известно точное выражение для Ψ, то энергия системы может быть рассчитана по формулам (6.13) или (6.14). Однако, обычно не известны ни Ψ ни Е либо неизвестна Ψ. Тогда для отыскания Ψ и Е можно воспользоваться вариационным принципом: подставив в (6.13) или (6.14) вместо истинной функции приближенную к ней так называемую пробную функцию Ψпробн, получим отвечающее ей значение Е. Оно обязательно будет не ниже энергии основного состояния системы E0
 . (6.15)
. (6.15)
http://spravochnick.ru/himiya/atomnye_i_molekulyarnye_orbitali/uravnenie_shredingera/
http://pandia.ru/text/78/418/15321-4.php