Поведение реальных газов описывает уравнение
Уравнение описывает поведение реальных газов при не слишком высоких давлениях и при достаточно высоких температурах. В действительности, при давлениях порядка 200 атм наблюдаются значительные отклонения от этого закона, которые, непрерывно возрастая с увеличением давления, достигают при 1000 атм более 100%. При выводе уравнения состояния идеального газа не принимались во внимание размеры молекул и их взаимодействие друг с другом. Между тем при повышении давления возрастает плотность газа, что приводит к уменьшению среднего расстояния между молекулами, вследствие чего размерами молекул и их взаимодействием пренебрегать уже нельзя. Размеры молекулы имеют порядок 10 -8 см. Тогда для объема одной молекулы получим:
Умножая эту величину на число Лошмидта (число молекул в 1 cм³), имеем объем молекул, находящихся в 1 cм³ при нормальных условиях:
Такой величиной действительно можно пренебречь по сравнению с объемом газа в 1 cм³. Однако, если увеличить давление до 5000 атм, то в 5000 раз должна возрасти плотность газа, и их объем в 1 cм³ составит уже 0,5 cм³. Доступным для движения молекул оказался бы объем, в два раза меньший, чем при нормальных условиях. Совершенно очевидно, что обратная пропорциональность объема газа давлению неизбежно должна нарушаться.
Самым простым и дающим хорошие результаты по писанию поведения реальных газов оказалось уравнение Ван-дер-Ваальса, полученное путем введения поправок в уравнение состояния идеального газа pVкм = RT:
где р — давление, оказываемое на газ извне и равное давлению газа на стенки сосуда, а и b — константы Ван-дер-Ваальса, имеющие для разных газов различные значения, определяемые опытным путем.
В системе СИ константа а измеряется в Н·м 4 /кмоль 2 , константа b — в м³/кмоль. Константа b определяет ту часть объема, которая недоступна для движения молекул вследствие их конечных размеров, и равна учетверенному объему молекулы. Пусть в сосуде находится только две молекулы. Центр любой из них не может приблизиться к центру другой на расстояние, меньшее диаметра молекулы d (Рис. 2.2.1).
Рис. 2.2.1. К объяснению физического смысла константы b
Таким образом, для центра этой молекулы оказывается недоступным сферический объем с радиусом d, т.е. объем, равный 8 объемам молекулы. Поскольку принято во внимание парное взаимодействие молекул, в расчете на одну молекулу недоступным оказывается объем, равный учетверенному объему молекулы. В газе молекулы взаимодействуют (сталкиваются) чаще всего попарно, поскольку вероятности столкновения трех и более молекул крайне малы, поэтому приведенное рассуждение справедливо для всего объема газа: константа b равна учетверенному суммарному объему всех молекул. Фактически введением константы b учитывают отталкивание между молекулами, возникающее при их сильном сближении.
Кроме сил отталкивания, между молекулами есть и силы притяжения. Поправка дает внутреннее давление pi, обусловленное взаимным притяжением молекул друг к другу. Если бы взаимодействие между молекулами вдруг прекратилось бы, то для того, чтобы удержать газ в пределах заданного объема потребовалось бы увеличить внешнее давление на величину pi. Обратная пропорциональность может быть объяснена так. Любая молекула, находящаяся вблизи стенки сосуда, будет иметь с одной стороны (обращенной внутрь сосуда) больше соседей, чем с другой. В результате эта молекула будет испытывать результирующую силу, направленную внутрь сосуда. Давление, которое испытывает пристенный слой молекул со стороны остальных молекул газа, равно силе f, действующей на все молекулы на единице поверхности слоя. Очевидно, что эта сила пропорциональна плотности молекул n. С другой стороны, число молекул N в пристенном слое, испытывающих притяжение, также пропорционально n. Следовательно, pi∼N·∼n 2 . Так как концентрация молекул обратно пропорциональна объему, занимаемому одним молем газа, то выполняется:
Коэффициент пропорциональности а должен быть измерен, поскольку нет способа точного его вычисления.
Чтобы перейти к произвольной массе газа, учтем, что z = m/μ киломолей газа при тех же условиях занимают в z раз больший объем: V = z·Vкм. Используя это обстоятельство, запишем уравнение (2.2.1) так:
Умножив это уравнение на z и вводя обозначения:
придем к уравнению Ван-дер-Ваальса для произвольной массы газа:
Насколько уравнение Ван-дер-Ваальса лучше описывает поведение реальных газов, чем уравнение состояния идеального газа, можно судить по данным для 1 л газообразного азота, занимающего при нормальных условиях объем 1 л при 0°С (Табл. 2.2.1).
| р, атм | pV, атм·л | , атм·л |
|---|---|---|
| 1 | 1,000 | 1,000 |
| 100 | 0,994 | 1,000 |
| 200 | 1,048 | 1,009 |
| 500 | 1,390 | 1,014 |
| 1000 | 2,069 | 0,893 |
Как видно из Табл. 2.2.1, уравнение Ван-дер-Ваальса гораздо лучше согласуется с экспериментом. Уравнение (2.2.5) можно представить так:
Получилось кубическое уравнение относительно неизвестной V, коэффициенты которого зависят от давления и температуры. Такое уравнение со свободным членом и вещественными коэффициентами имеет три решения, причем в зависимости от соотношения между коэффициентами либо все три решения будут вещественными, либо одно решение — вещественным, а два других — комплексными. Поскольку объем может быть только вещественной величиной, комплексные решения не имеют физического смысла. На Рис. 2.2.2 показаны изотермы Ван-дер-Ваальса для нескольких значений температур.
Рис. 2.2.2. Изотермы Ван-дер-Ваальса
При температуре Т’ и давлениях в пределах p’1 до p’2 коэффициенты в уравнении (2.2.6) таковы, что все три решения оказываются вещественными; при иных давлениях вещественным будет только одно решение. Начиная с определенной, своей для каждого вещества температуры Ткр ( критической температуры ) при любом давлении вещественным остается только одно решение уравнения (2.2.6). Если повышать температуру, то точки, соответствующие решениям уравнения V’1, V’2, V’3, все больше сближаются, сливаясь при критической температуре в одну точку К, также называемую критической точкой . Для соответствующей изотермы точка К является точкой перегиба. Ей соответствуют три совпадающих вещественных решения уравнения (2.2.6). Касательная к критической изотерме в точке К будет параллельна оси V, так что в этом случае производная . Кроме того, в точке перегиба должна быть равна нулю и вторая производная .
Разрешим уравнение (2.2.1) относительно р:
Дифференцирование (2.2.7) по объему дает:
| (2.2.8) |
| (2.2.9) |
В критической точке, т.е. при подстановке Т = Ткр, Vкм = Vкм.кр, эти выражения должны обращаться в нуль:
| (2.2.10) |
| (2.2.11) |
Совместно с (2.2.7), записанным для точки К:
они образуют три уравнения с тремя неизвестными ркр, Vкм.кр и Ткр. Решение этой системы уравнений дает:
Таким образом, зная константы Ван-дер-Ваальса а и b, можно найти соответствующие критической точке параметры ркр, Vкм.кр и Ткр, которые называются критическими величинами . И, наоборот, по известным критическим величинам могут быть найдены значения констант Ван-дер-Ваальса. Из (2.2.13) и (2.2.12) можно получить:
в то время как согласно уравнению состояния идеального газа должно было бы выполняться соотношение:
2.2.2. Внутренняя энергия реального газа
Взаимодействие между молекулами реального газа обусловливает их взаимную потенциальную энергию, которая должна учитываться во внутренней энергии газа наряду с кинетической энергией движения молекул:
Кинетическая энергия киломоля газа равна:
и прямо пропорционально зависит от температуры.
Потенциальная энергия взаимодействия молекул зависит от среднего расстояния между ними, поэтому Еп должна быть функцией объема газа V. Следовательно, внутренняя энергия газа есть функция двух параметров — температуры и объема:
При расширении газа совершается работа по преодолению сил притяжения между молекулами. Работа против внутренних сил, действующих между молекулами киломоля газа, может быть записана в виде:
Приравнивая (2.2.19) приращению потенциальной энергии, получим:
Интегрируя (2.2.20), имеем:
Значение постоянной интегрирования полагают равной нулю, что при увеличении объема газа до бесконечности соответствует отсутствию взаимодействия между молекулами газа. Тогда полное выражение для внутренней энергии киломоля реального газа будет иметь вид:
Из (2.2.22) следует, что внутренняя энергия растет как при повышении температуры, так и при увеличении объема.
Если газ будет расширяться или сжиматься без теплообмена с внешней средой и без совершения над ним внешней работы, то, согласно первому началу термодинамики, его внутренняя энергия должна оставаться постоянной. Тогда из (2.2.22) получим:
из чего следует, что приращения dT и dVкм имеют противоположный знак.
Следовательно, при расширении в таких условиях газ всегда должен охлаждаться, а при сжатии — нагреваться.
© ФГОУ ВПО Красноярский государственный аграрный университет, 2015
Уравнение состояния вещества
Параметры состояния связаны друг с другом. Соотношение, при котором определяется данная связь, называют уравнением состояния данного тела. В самом простом случае равновесное состояние тела определяется значением следующих параметров: давления p , объема V и температуры (масса тела или системы, как правило, известна).
Что такое идеальный газ
Уравнение состояния так называемого идеального газа является простым, но достаточно информативным.
Идеальный газ – это газ, в котором пренебрегают взаимодействием молекул между собой.
Идеальными считают разреженные газы. Особенно близки к идеальным газы гелий и водород. Идеальный газ – это упрощенная математическая модель реального газа: молекулы движутся хаотически, а соударения между молекулами и удары молекул о стенки сосуда упругие, не приводящие к потерям энергии в системе. Подобная упрощенная модель весьма удобна, поскольку не требует учета силы взаимодействия между молекулами газа. Множество реальных газов не отличаются в своем поведении от идеального газа в условиях, когда суммарный объем молекул пренебрежимо мал в сравнении с объемом сосуда (то есть при атмосферном давлении и комнатной температуре). Это дает возможность применять уравнение состояния идеального газа для сложных расчетов.
Уравнение состояния идеального газа запишем несколько раз ( 2 ) , ( 3 ) , ( 5 ) :
p V = m μ R T = ν R T ( 2 ) .
Уравнение ( 2 ) – уравнение Менделеева-Клапейрона, где m – это масса газа, μ – это молярная масса газа, R = 8 , 31 Д ж м о л ь · К – это универсальная газовая постоянная, ν – это число молей вещества.
где N – это количество молекул газа в массе m , k = 1 , 38 · 10 — 23 Д ж К , постоянная Больцмана, определяющая «долю» газовой постоянной, которая приходится на 1 молекулу и
N A = 6 , 02 · 10 23 м о л ь — 1 – это постоянная Авогадро.
Если поделить в ( 4 ) обе части на V , то получаем следующий вид записи уравнения состояния идеального газа:
где n = N V – это количество частиц в единице объема или же концентрация частиц.
Что такое реальный газ
Рассмотрим теперь более сложные системы: неидеальные газы и жидкости.
Реальный газ – это газ, между молекулами которого наблюдаются заметные силы взаимодействия.
Необходимо учитывать, что в неидеальных, плотных газах взаимодействие молекул высоко. Известно, что взаимодействие молекул очень сильно усложняет физическую картину, потому точную формулу уравнения состояния неидеального газа не получается записать в простом виде. В данном случае прибегают к приближенным формулам, найденным полу-эмпирическим путем. Самая удачная формула – это уравнение Ван-деp-Ваальса.
Взаимодействие молекул обладает сложным характером. На достаточно больших расстояниях между молекулами действуют силы притяжения. С уменьшением расстояния силы притяжения вначале растут, однако потом уменьшаются и преобразуются в силы отталкивания. Притяжение и отталкивание молекул будем рассматривать и учитывать отдельно. Уравнение Ван-дер-Ваальса, которое описывает состояние одного моля реального газа, имеет вид:
p + a V μ 2 V μ — b = R T ( 6 ) ,
где a V μ 2 – это внутреннее давление, обусловленное силами притяжения между молекулами, b – это поправка на собственный объем молекул, учитывающая действие сил отталкивания между молекулами, при этом:
b = N A 2 3 πd 3 ( 7 ) ,
где d – это диаметр молекулы. Значение a рассчитывается по формуле:
a = — 2 πN A 2 ∫ d ∞ W p ( r ) r 2 dr ( 8 ) ,
где W p ( r ) – это потенциальная энергия притяжения 2 -х молекул.
При увеличении объема значение поправок в уравнении ( 6 ) становится менее существенным. И в пределе уравнение ( 6 ) превращается в уравнение ( 2 ) . Это согласовано с тем фактом, что с уменьшением плотности реальные газы по своим характеристикам приближаются к идеальным.
Положительным в уравнении Ван-деp-Ваальса является тот факт, что данное равенство при очень больших плотностях приблизительно описывает также и свойства жидкости, в частности, плохую ее сжимаемость. Потому существует основание предполагать, что уравнение Ван-деp-Ваальса позволяет отразить и переход от жидкости к газу (либо от газа к жидкости).
На рисунке 1 представлена изотерма Ван-дер-Ваальса для некоторого постоянного значения температуры T , которая построена из соответствующего уравнения.
В месте “извилины” (участок КМ) изотерма 3 раза пересекает изобару. На участке
V 1 , V 2 давление увеличивается с ростом объема.
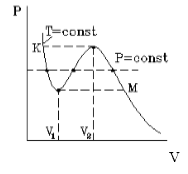
Подобная зависимость невозможна. Это означает, что в этой области с веществом происходит что-то необыкновенное. Что именно, не видно в уравнении Ван-деp-Ваальса. Обратимся к опыту. В месте “извилины” на изотерме в состоянии равновесия вещество расслаивается на 2 фазы: жидкую и газообразную. Обе фазы существуют одновременно и находятся в фазовом равновесии. В таком состоянии происходит испарение жидкости и конденсация газа. Процессы протекают с такой интенсивностью, что полностью компенсируют друг друга: объем жидкости и газа со временем не изменяется.
Газ, который находится в фазовом равновесии со своей жидкостью, называется насыщенным паром. Если фазовое равновесие отсутствует, отсутствует также компенсация испарения и конденсации, тогда газ называется ненасыщенным паром.
Что происходит с изотермой в области двухфазного состояния вещества (то есть в месте «извилины» изотермы Ван-деp-Ваальса)? Эксперимент показывает, что в этом месте при изменении объема давление остается неизменным. График изотермы идет параллельно оси V (рисунок 2 ).
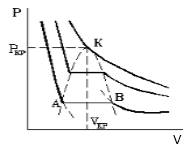
С увеличением температуры участок двухфазных состояний на изотермах уменьшается до тех пор, пока не превращается в точку (рисунок 2 ). Это особая точка К , в которой исчезает разница между жидкостью и паром. Ее называют критической точкой.
Параметры, которые соответствуют критическому состоянию, являются критическими (критическая температура, критическое давление, критическая плотность вещества).
Необходимо найти величину n . В процессе, представленном на рисунке 3 , давление p
Из графика, приведенного на рисунке 3 , запишем уравнение процесса в параметрах p ( V ) :
С учетом уравнения Менделеева-Клайперона:
V вместо объема, получаем:
Моль кислорода охлаждают до — 100 ° C . Необходимо определить давление, которое оказывает газ на стенки сосуда, если занимаемый газом объем V = 0 , 1 л . Необходимо также сравнить p с давлением идеального газа p i d , если бы кислород вел себя как идеальный газ. Величина постоянных Ван-дер-Ваальса a и b , для кислорода a = 0 , 1358 П а · м 6 / м о л ь 2 , b = 3 , 167 · 10 — 5 м 3 / м о л ь .
Из уравнения Ван-Дер-Ваальса имеем:
p = R T V μ — b — a V μ 2
Переведем температуру в систему измерения: T = t + 273 , По условию T = 173 K , V = 0 , 1 л = 10 — 4 м 3 .
Произведем расчет: p = 8 , 31 · 173 ( 10 — 3 , 2 ) · 10 — 5 — 0 , 1358 ( 10 — 4 ) 2 = 75 , 61 · 10 5 ( П а ) .
Для идеального газа:
Рассчитаем: p i d = 1 · 8 , 31 · 173 10 — 4 = 143 · 10 5 ( П а ) .
Уравнения состояния реальных газов
Все реальные газы являются парами тех или иных жидкостей, причем, чем ближе газ к переходу в жидкое состояние, тем больше его отклонение от свойств идеального газа, состояние которого описывается уравнением Клапейрона. Для качественной оценки особенностей реальных газов рассмотрим область, где будут значительные отступления от уравнения, описывающего поведение идеальных газов.
Если сжимать газ при постоянной температуре, то можно достигнуть состояния насыщения (сжижения газа), соответствующего этой температуре и некоторому определенному давлению. При дальнейшем сжатии пар будет конденсироваться и в определенный момент полностью превратится в жидкость.
Процесс перехода пара в жидкость проходит при постоянных температуре и давлении, так как давление насыщенного пара однозначно определяется температурой. На р – u -диаграмме (рисунок 10.1) область двухфазных состояний (пар и жидкость) лежит между кривыми кипящей жидкости и сухого насыщенного пара. При увеличении давления эти кривые сближаются. Сближение происходит потому, что объем пара, уменьшается, а объем жидкости увеличивается.
При некотором определенном для данной жидкости (пара) давлении кривые кипящей жидкости и пара встречаются в так называемой критической точке, которой соответствуют критические параметры: давление ркр, температура Ткр, удельный объем uкр, характеризующие критическое состояние вещества.
При критическом состоянии исчезают различия между жидкостью и паром. Оно является предельным физическим состоянием, как для однородного, так и для распавшегося на две фазы вещества.
При температуре более высокой, чем критическая, газ ни при каком давлении не может сконденсироваться, т. е. превратиться в жидкость.
В общем случае все газы в области, близкой к состоянию сжижения, приближенно воспроизводят связь между параметрами состояния по уравнению Клапейрона. Во всех газах с более или менее значительной плотностью нельзя пренебрегать силами сцепления между молекулами, объемом, занимаемым ими, а также ассоциацией молекул в группы.
 Под ассоциацией понимается механическое соединение двух или нескольких молекул в одну сложную. Уменьшение числа самостоятельных частиц, из которых состоит газ, должно привести к возрастанию среднего молекулярного веса газа и уменьшению его давления. Ассоциация значительно усложняет математическое описание состояния реальных газов.
Под ассоциацией понимается механическое соединение двух или нескольких молекул в одну сложную. Уменьшение числа самостоятельных частиц, из которых состоит газ, должно привести к возрастанию среднего молекулярного веса газа и уменьшению его давления. Ассоциация значительно усложняет математическое описание состояния реальных газов.
|
|
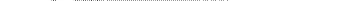 При уменьшении давления и возрастании температуры газа можно пользоваться уравнением состояния идеального газа за иск точением тех случаев, когда в газе под влиянием больших температур наступает изменение химической структуры (например, распад сложных молекул — диссоциация).
При уменьшении давления и возрастании температуры газа можно пользоваться уравнением состояния идеального газа за иск точением тех случаев, когда в газе под влиянием больших температур наступает изменение химической структуры (например, распад сложных молекул — диссоциация).
Уравнение состояния реальных газов выводится или чисто теоретически на основе гипотетических представлений о структуре газа, или на основании обработки экспериментальных зависимостей между р, u, Т.
Широкое распространение в научных исследованиях получило уравнение Ван-дер-Ваальса, выведенное путем пересмотра некоторых допущений, лежащих в основе уравнения состояния идеального газа. Уравнение состояния реального газа с учетом сил, действующих между молекулами, и их объема для 1 кг газа имеет вид
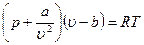 (10.1)
(10.1)
Это уравнение отличается от уравнения Клапейрона двумя поправками: поправкой на объем самих молекул b и поправкой на так называемое внутреннее давление — 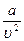 определяемое взаимным притяжением молекул газа. Это давление может рассматриваться как сила, действующая со стороны внешних периферийных молекул и направленная внутрь сосуда.
определяемое взаимным притяжением молекул газа. Это давление может рассматриваться как сила, действующая со стороны внешних периферийных молекул и направленная внутрь сосуда.
Рассмотрим изменения на изотермах, обусловленных поправками а и b. При температуре выше критической изотермы, построенные по уравнению Ван-дер-Ваальса, представляют собой плавные кривые, отличные от равнобоких гипербол, которые бы дало уравнение состояния идеального газа. Последние в верхней части на рисунка 9.2 показаны пунктиром.
При температуре ниже критической имеется область объемов, где поправка (уменьшающая давление) играет определяющую роль и давление проходит через максимум в точке С. Для меньших объемов давление падает, проходит через минимум — точка В, а затем резко увеличивается, когда u стремится к значению b.
При критической температуре, максимум и минимум на изотермах сливаются в точке перегиба К, а так как касательная к изотерме в критической точке идет горизонтально, то для критической точки должны выполняться условия
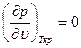 ,
, 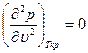 . (10.2)
. (10.2)
Отсюда получаем уравнения
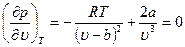 , (10.3)
, (10.3)
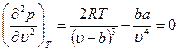 . (10.4)
. (10.4)
Температуру Ткр и объем uкр можно определить из уравнений (10.3) и (10.4), а давление ркр находится затем из уравнения (10.1). В результате получаем
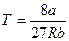 ,
, 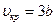 ,
,  (10.5)
(10.5)
Из последних соотношений можно определить индивидуальные константы а и b, зависящие от физических свойств данного газа
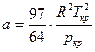 ,
, 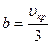 .
.
Так как процесс перехода от жидкого состояния к газообразному идет при постоянных Т и р, то на рисунке 10.2 этому процессу соответствует линия АD.
Однако участки АВ и СD на изотермах можно воспроизвести экспериментально только при использовании очень чистых жидкостей и газов. Вещество на этих участках находится в виде перегретой жидкости и перенасыщенного (переохлажденного) пара. Такие состояния, когда вещество остается в однофазном состоянии и не распадается на фазы, называются метастабильными.
Главная ценность уравнения Ван-дер-Ваальса состоит в том, что оно качественно правильно описывает непрерывность перехода из жидкого состояния в газообразное и дальнейшее развитие уравнения состояния пошло по пути уточнения расчетов и усовершенствования его теории.
Предпринимались попытки усовершенствования его за счет того, что коэффициенты а и b принимались не постоянными, а зависящими от температуры и объема. Но эти попытки не привели к созданию уравнения состояния, описывающего свойства газа в широком диапазоне изменения параметров.
Неудачи создания общего уравнения состояния привели к появлению целого ряда эмпирических уравнений, которые могли бы с достаточной точностью предсказывать поведение реальных газов в широком диапазоне условий. Наиболее известны из них: уравнение Битти – Бриджмена с пятью эмпирически определяемыми постоянными и уравнение Бенедикта-Вебб-Рубина, содержащее восемь эмпирических постоянных – (а, b, с, d, А0, В0, С0 и т.д.)
Уравнение Битти – Бриджмена, применяемое до давлений порядка 250 бар и плотностей газа, не превышающих 0,5 плотности в критической точке, имеет вид
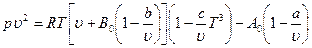 , (10.7)
, (10.7)
а уравнение состояния Бенедикта – Вебб – Рубина имеет вид
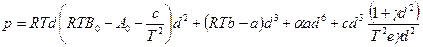 (10.8)
(10.8)
Эти уравнения могут предсказать р, u, Т – свойства газа с ошибкой в пределах нескольких десятых процента и, несмотря на их сложность, развитие вычислительной техники стимулирует использование таких уравнений состояния в обычных технических расчетах.
Хорошо согласуется с опытными данными одно из современных уравнений состояния газа — уравнение Вукаловича — Новикова, учитывающее ассоциацию молекул. При учете столкновений двойных молекул это уравнение имеет вид
 , (10.9)
, (10.9)
где 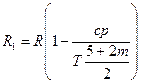 ,
,
с и m — опытные константы.
В настоящее время теоретически обосновано уравнение состояния, представляющее собой разложение коэффициента сжимаемости z в бесконечный ряд по степеням 1/u
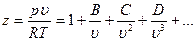 (10.10)
(10.10)
где В, С и D — второй, третий и четвертый вириальные коэффициенты, учитывающие взаимодействие соответственно двух трех, четырех и т. д. молекул. Вириальные коэффициенты зависят лишь от температуры и определяются, если известна зависимость потенциальной энергии взаимодействия молекул U от расстояния между ними (рисунок 10.3).
Вместо точных аналитических зависимостей Uпот= f(r) практически применяют приближенные выражения, которые называются потенциалами. Широко используется потенциал Леннарда — Джонса, по которому энергия отталкивания пропорциональна двенадцатой степени расстояния между молекулами, а энергия притяжения – шестой
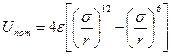 , (10.11)
, (10.11)
где r — расстояние между молекулами; s— значение r, при котором Uпот = 0; e – максимальная величина энергии притяжения (глубина потенциальной ямы).
 Значения s и e для каждого газа могут быть определены по экспериментальным данным. Кроме потенциала Леннарда – Джонса применяются другие потенциалы, которые могут быть использованы, для определенных групп сходственных веществ.
Значения s и e для каждого газа могут быть определены по экспериментальным данным. Кроме потенциала Леннарда – Джонса применяются другие потенциалы, которые могут быть использованы, для определенных групп сходственных веществ.
|
При решении целого ряда технических задач рабочими телами могут быть не широко используемые в технике вещества (водяной пар, углекислый газ, азот и некоторые другие), а вещества, термические свойства которых неизвестны.
В этом случае можно воспользоваться для предсказания свойств малоизученных веществ положением о термодинамическом подобии веществ.
Если значения индивидуальных констант а и b подставить в уравнение (10.1), то получим уравнение Ван-дер-Ваальса в функции приведенных параметров
 , (10.12)
, (10.12)
где 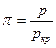 ,
,  ,
, 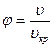 .
.
Эти отношения называются приведенными давлением, температурой и объемом. Уравнение (10.12) можно записать в форме
 (10.13)
(10.13)
В этой форме приведенное уравнение состояния будет одинаково для всех веществ. Состояния двух или нескольких веществ, в которых они имеют одинаковые приведенные параметры 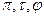 , называются соответственными состояниями, т. е. эти вещества находятся в состояниях, пропорционально удаленных от своего критического состояния.
, называются соответственными состояниями, т. е. эти вещества находятся в состояниях, пропорционально удаленных от своего критического состояния.
Если вещества подчиняются одному и тому же приведенному уравнению состояния и имеют два одинаковых приведенных параметра, то у них одинаков и третий приведенный параметр, т. е. вещества, будут находиться в соответственных состояниях. Это положение носит название закона соответственных состояний.
Вещества, подчиняющиеся закону соответственных состояний, называют термодинамически подобными.
Практически закон соответственных состояний наиболее удобно применять в виде зависимости 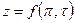 , причем для расчетов можно применить
, причем для расчетов можно применить 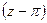 — диаграмму (рисунок 10.4). Эта диаграмма строится по экспериментальным данным дляразличных веществ и может быть использована для расчета термодинамических свойств малоизученных веществ методом термодинамического подобия.
— диаграмму (рисунок 10.4). Эта диаграмма строится по экспериментальным данным дляразличных веществ и может быть использована для расчета термодинамических свойств малоизученных веществ методом термодинамического подобия.
Для этого, зная критические параметры вещества, находят и , а по 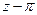 -диаграмме определяется коэффициент сжимаемости при данных приведенных параметрах. Значение удельного объема можно вычислить по формуле
-диаграмме определяется коэффициент сжимаемости при данных приведенных параметрах. Значение удельного объема можно вычислить по формуле
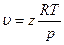 .
.
Точность расчета по этому методу не превышает 15%, так как закон соответственных состояний выполняется лишь приближенно. Так, при одинаковых я и т коэффициенты сжимаемости должны быть равны, причем должны быть равны и коэффициенты сжимаемости в критической точке 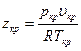 . Но для реальных веществ
. Но для реальных веществ 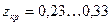 , следовательно, строго говорить о выполнении закона соответственных состояний можно лишь для узких групп сходственных между собой веществ.
, следовательно, строго говорить о выполнении закона соответственных состояний можно лишь для узких групп сходственных между собой веществ.
Парообразование при постоянном давлении
Рассмотрим изменение состояния водяного пара (реального газа), имеющего сравнительно высокую критическую температуру. Изменение параметров состояния водяного пара удобно проследить на р-u -диаграмме (рис. 9.5).
Положим, что 1 кг воды при 0° С заключен в цилиндре, закрытом свободно движущимся невесомым поршнем, на который действует постоянное внешнее давление. Объем воды при указанных условиях обозначим 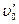 . Если считать жидкость несжимаемой при любых давлениях, то прямая, соединяющая точки l¢, l, l² и параллельная оси ординат, будет определять все возможные состояния воды при 0° С. Следует отметить, что для несжимаемой жидкости эта изохора совпадает с изотермой.
. Если считать жидкость несжимаемой при любых давлениях, то прямая, соединяющая точки l¢, l, l² и параллельная оси ординат, будет определять все возможные состояния воды при 0° С. Следует отметить, что для несжимаемой жидкости эта изохора совпадает с изотермой.
Если (при постоянном давлении) подводить к жидкости теплоту, то при достижении температуры кипения tВ начнется превращение воды в пар — точка т. Удельный объем жидкости вследствие нагрева увеличивается от до 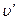 . При более высоком давлении процесс парообразования начнется и при более высокой температуре tн, следовательно, объем воды при достижении точки кипения будет больше, чем раньше (точка т).
. При более высоком давлении процесс парообразования начнется и при более высокой температуре tн, следовательно, объем воды при достижении точки кипения будет больше, чем раньше (точка т).
На р-u -диаграмме геометрическое место точек, определяющих состояние воды, нагретой до температуры кипения, изображается кривой т¢, т, т². Эту кривую называют нижней (левой) пограничной кривой. При дальнейшем подведении теплоты начинается процесс парообразования. При условии постоянства давления, как показывает опыт, для всех жидкостей имеет место характерное явление: температура смеси жидкости и пара остается неизменной и равной температуре кипения tH.
 Процесс парообразования прекратится в точке n, когда вся жидкость превратится в пар. Между точками т и п система — двухфазная, пар в этой области — влажный насыщенный.
Процесс парообразования прекратится в точке n, когда вся жидкость превратится в пар. Между точками т и п система — двухфазная, пар в этой области — влажный насыщенный.
Влажный насыщенный пар представляет собой смесь пара с жидкостью, причем жидкость может быть сосредоточена в нижней части цилиндра или равномерно распределена в виде мельчайших капель по всему объему.
Пар, полученный при испарении всей жидкости (точка п), — сухой насыщенный. Удельный объем, пара в этой точке обозначим через u». При проведении процесса парообразования при другом давлении соответственно получим точки n¢, п». Кривая п’ п п» представляет собой верхнюю (правую) пограничную кривую. Пересечение верхней и нижней пограничных кривых определяет положение критической точки К. Для воды критической точке соответствует ркр = 221,048 бар, Ткр = 647,15° К; uкр = 0,0031 м 3 /кг. На рис. 9.5 в области влажного насыщенного пара пунктирными линиями показаны линии постоянной сухости. Степень сухости пара х представляет собой массовую долю сухого насыщенного пара во влажном
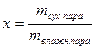 (9.14)
(9.14)
Для точек, лежащих на нижней пограничной кривой, х = 0, для точек, лежащих на верхней пограничной кривой, х = 1. Если к сухому насыщенному пару продолжать подводить теплоту, то удельный объем и температура увеличиваются (un > u», t > tн). Пар в этом состоянии называют перегретым. Начиная с точки п вправо система однофазная.
Изменение агрегатного состояния
 В § 2 рассматривался процесс парообразования, т. е. переход из жидкого состояния в парообразное, осуществляемый при постоянном давлении. Аналогичный переход из твердого состояния в газообразное называют возгонкой, или сублимацией, а из твердого состояния к жидкому — плавлением.
В § 2 рассматривался процесс парообразования, т. е. переход из жидкого состояния в парообразное, осуществляемый при постоянном давлении. Аналогичный переход из твердого состояния в газообразное называют возгонкой, или сублимацией, а из твердого состояния к жидкому — плавлением.
Состояния вещества при этих превращениях считают устойчивыми, стабильными. При этом всякие изменения состояния считаются квазистатическими, как это обычно принято в термодинамике.
Переход из одного агрегатного состояния в другое удобно рассматривать на р – t — диаграмме (рис. 9.6). На диаграмме кривая АК представляет собой зависимость между давлением насыщенного пара и температурой кипения, т. е. р = f (tн) (кривая упругости пара).
Кривая равновесия жидкой и газообразной фазы заканчивается в критической точке К.
Если от жидкости отбирать теплоту при постоянном давлении, то при определенной температуре жидкость переходит в твердое состояние. Температура, при которой осуществляется этот переход, называется температурой затвердевания, или плавления tпл, а количество теплоты, отбираемое в этом процессе, называется скрытой теплотой плавления. При плавлении так же, как и при парообразовании, вещество находится в двух фазах. Аналогично кривой АК можно построить кривую AD, которая однозначно определяется зависимостью р = f(tпл).
Кривая сублимации АВ представляет собой зависимость р = f(tc) для перехода твердого тела в газообразное. Этот переход при температуре сублимации tc происходит вследствие подведения некоторого количества теплоты, носящего название скрытой теплоты сублимации. Точки этой кривой соответствуют двухфазной системе твердое тело — газ (например, водяной пар над поверхностью льда).
Все три кривых равновесия (парообразования, плавления и сублимации) пересекаются в некоторой характерной для каждого вещества точке. Эта точка А называется тройной точкой, а изображаемое ею состояние — фундаментальным. В этой точке находятся в термодинамическом равновесии три различные фазы вещества: твердая, жидкая и газообразная.
Тройной точке воды соответствуют следующие параметры: давление р = 0,00610 бар, Т =273,16°К.
Рассмотрение описанных процессов показывает, что в состояниях, находящихся между кривыми АВ, АЕ и AD, тело будет находиться целиком в одной фазе: правее АВ и АК — область газообразного состояния; левее линий AD и АВ располагается область вещества в твердом состоянии; между линиями AD и АК находится область жидкости.
В состояниях на линии АК, AD и АВ вещество может существовать в двух фазах, причем на линии АК в жидкой и газообразной, на AD —твердой и жидкой; а на линии АВ вещество может быть в твердом и газообразном состояниях. Расположение и вид этих трех кривых
зависят от природы вещества и устанавливаются опытным путем.
Параметры состояния воды и водяного пара
Вследствие незначительной сжимаемости воды можно принять, что плотность воды при 0° С и любых давлениях есть величина постоянная, a u’0 = 0,001 м 3 /кг. Начало отсчета внутренней энергии энтальпии и энтропии берется от 0° С и соответствующего давления насыщения р = 0,00610 бар. При этих параметрах энтальпия, энтропия, а также внутренняя энергия воды берутся условно равными нулю: s’0 = 0, i’0 = 0, и’0 = 0.
В процессе подогрева воды происходит нагревание ее до температуры кипения tн. Удельный объем воды при температуре кипения u’ будет больше объема u’0. Соответствующие значения u’ для воды в функции температуры и давления для состояний, лежащих или на нижней пограничной кривой, или левее ее, даются в справочной литературе.
Количество теплоты, которое нужно сообщать воде, чтобы нагреть ее от 0° С до температуры кипения в процессе р = const, называется теплотой жидкости. Это количество теплоты определяется по формуле
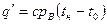 , (9.15)
, (9.15)
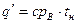 (9.16)
(9.16)
где 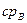 — средняя теплоемкость воды в интервале температур от 0° С до tН°С
— средняя теплоемкость воды в интервале температур от 0° С до tН°С
При низких по сравнению с Ткр температурах можно считать = 4,1865 кдж/(кг·град).
Воспользуемся в изобарном процессе подогрева воды первым за’ коном термодинамики, по которому
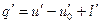 (9.17)
(9.17)
где и’ — внутренняя энергия воды при температуре кипения.
Так как при 0° С и¢0 = 0, а работа расширения жидкости
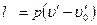 (9.18)
(9.18)
практически заметна только при больших значениях давления, то
 (9.19)
(9.19)
Энтальпия воды при температуре кипения определяется по общей формуле
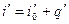 (9.20)
(9.20)
Полагая, что 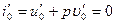 , получим
, получим
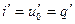 (9.21)
(9.21)
В процессе нагревания жидкости от 0° С до температуры кипения происходит увеличение ее энтропии, которое может быть найдено по формуле
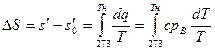 (9.22)
(9.22)
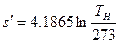 (9.23)
(9.23)
Как уже было сказано, опытами установлено, что в процессе парообразования жидкость, нагретая до температуры кипения при . этой температуре и определенном постоянном давлении, обращается в пар. Количество теплоты, затрачиваемое в процессе при р = const на превращение 1 кг воды при температуре кипения в сухой насыщенный пар той же температуры, обозначим через г.
Теплота г называется скрытой теплотой парообразования. По первому закону термодинамики
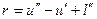 (9.24)
(9.24)
где и²— внутренняя энергия сухого насыщенного пара;
l» — работа расширения в процессе парообразования.
Разность внутренних энергий и» — и¢ затрачиваемая на работу против внутренних сил, называется внутренней теплотой парообразования и обозначается буквой r. Теплота, затрачиваемая на работу против внешних сил, равна
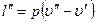 (9.25)
(9.25)
и называется внешней теплотой парообразования. Обозначим ее буквой y.
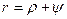 (9.26)
(9.26)
Вследствие того, что процесс парообразования идет при постоянном давлении,
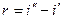 (9.27)
(9.27)
Величины r и i» даются в таблицах насыщенного пара, а 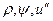 — легко определяются по приведенным выше формулам.
— легко определяются по приведенным выше формулам.
С возрастанием давления, как видно из рис. 9.7, увеличивается энтальпия жидкости и достигает максимального значения при критическом давлении. Скрытая теплота парообразования уменьшается с ростом давления и равна нулю при критическом давлении (и температуре), потому что в этих условиях различия между жидкостью и ее паром исчезают и процесс парообразования как таковой отсутствует.
Изменение энтропии в процессе парообразования при подведении к кипящей воде r кдж/кг теплоты равно
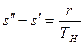 (9.28)
(9.28)
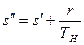 (9.29)
(9.29)
или, используя значение s¢ из выражения (9.23),
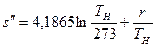 (9.30)
(9.30)
 При полном испарении жидкости состояние сухого насыщенного пара определяется одним параметром: давлением или температурой. Поэтому объем, внутренняя энергия и энтальпия определяются по таблицам насыщенного пара по давлению или температуре.
При полном испарении жидкости состояние сухого насыщенного пара определяется одним параметром: давлением или температурой. Поэтому объем, внутренняя энергия и энтальпия определяются по таблицам насыщенного пара по давлению или температуре.
Связь между удельными объемами жидкости и пара на линии насыщения u¢ и u² давлением насыщенного пара рН температурой ТН и скрытой теплотой парообразования может быть получена следующим образом. При превращении жидкости в пар давление насыщенного пара от объема системы не зависит, следовательно, в выражении (8.8)  , но так как равновесное превращение жидкости в пар происходит при постоянной температуре (ТН=const), то
, но так как равновесное превращение жидкости в пар происходит при постоянной температуре (ТН=const), то
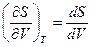
где dV представляет изменение объема системы при переходе жидкости в пар. Таким образом,
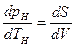 (9.31)
(9.31)
Изменение объема системы, если испарилась жидкость массой dm, равно
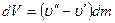
а приращение энтропии в квазистатическом процессе испарения жидкости массой dm по (9.28)
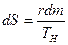
Подставив эти значения в уравнение (9.31), получим
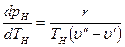 (9.32)
(9.32)
где 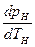 — производная от давления по температуре на кривой фазового равновесия рН = f (TН).
— производная от давления по температуре на кривой фазового равновесия рН = f (TН).
Уравнение (9.32) называют уравнением Клапейрона—Клаузиуса и применяют при исследованиях изменений агрегатного состояния вещества из жидкого состояния в парообразное. Аналогичные уравнения можно применять и к процессам перехода вещества из твердого состояния в жидкое или газообразное.
Параметры влажного насыщенного пара при заданной величине сухости могут быть определены из следующих соотношений.
Удельный объем влажного насыщенного пара
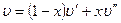 (9.33)
(9.33)
Так как объем воды (1 — х) 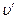 мал по сравнению с объемом пара, то при невысоких давлениях
мал по сравнению с объемом пара, то при невысоких давлениях
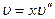 (9.34)
(9.34)
Энтальпия влажного насыщенного пара с учетом того, что на превращение в пар х кг жидкости необходимо затратить хr кдж/кг теплоты, равна
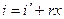 (9.35)
(9.35)
Энтропия влажного насыщенного пара
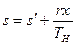 (9.36)
(9.36)
Свойства перегретого пара резко отличаются от свойств насыщенного пара и приближаются к свойствам газов.
Перегретый пар характеризуется тем, что его температура выше температуры парообразования ТH при том же давлении и удельный объем его больше, чем объем сухого насыщенного пара при том же давлении.
Количество теплоты, необходимое для перевода 1 кг сухого насыщенного пара при р = const в перегретый с температурой t, называют теплотой перегрева qпи определяют по формуле
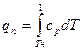 (9.37)
(9.37)
Если срm — средняя массовая теплоемкость перегретого пара при постоянном давлении, то
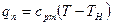 (9.38)
(9.38)
Значение срm берется для перегретого пара по формуле 
Энтальпия перегретого пара
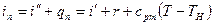 (9.39)
(9.39)
называется полной теплотой перегретого пара. По первому закону термодинамики
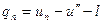 (9.40)
(9.40)
где 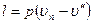 — работа расширения в изобарном процессе перегрева пара;
— работа расширения в изобарном процессе перегрева пара;
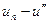 — изменение внутренней энергии в процессе перегрева.
— изменение внутренней энергии в процессе перегрева.
Изменение энтропии в равновесном изобарном процессе перегрева равно
 (9.41)
(9.41)
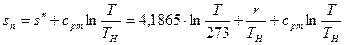 (9.42)
(9.42)
Свойства перегретых паров будут тем ближе к свойствам идеального газа, чем больше температура перегрева.
Т—s-диаграмма водяного пара
Для графического изображения процессов, происходящих в паре, удобно пользоваться Т — s-диаграммой, ибо в ней площадь под кривой обратимого процесса дает количество теплоты, сообщаемое телу или отнимаемое от него. Так как в системах координат р — v и Т—s любая точка изображает определенное состояние тела, то точкам р- 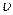 диаграммы должны соответствовать определенные точки, Т—s диаграммы (рис. 9.8).
диаграммы должны соответствовать определенные точки, Т—s диаграммы (рис. 9.8).
Если было принято условно, что энтропия начального состояния воды so = 0, то эта точка лежит на оси ординат на 273° выше абсолютного нуля.
Перенося по точкам нижнюю пограничную кривую (х = 0) из системы р — v в Т — s-диаграмму, получим соответствующую ей кривую, абсциссами которой являются значения s’. Аналогично наносится верхняя пограничная кривая (х = 1), абсциссами которой будут значения энтропии сухого насыщенного пара s».
В точке b диаграммы начинается кипение при ТH = const, и энтропии в процессе парообразования повышается
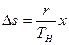
Процесс парообразования заканчивается в точке с, где
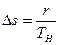
Так как процесс парообразования идет при Тн = const и р — =const, изотерма b-с является одновременно и изобарой. Дальнейший подвод теплоты снова сопровождается увеличением температуры и энтропии. В процессе перегрева пара (кривая с-е)
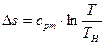
 Вследствие того что площади в Т — s-диаграмме изображают количество подведенной (отведенной) теплоты, то пл. аbАО — теплота в процессе нагрева жидкости от 0° С до температуры кипения; пл. abА0 — теплота, подводимая к воде в процессе парообразования; пл. сеСВ — теплота, затраченная на перегрев пара.
Вследствие того что площади в Т — s-диаграмме изображают количество подведенной (отведенной) теплоты, то пл. аbАО — теплота в процессе нагрева жидкости от 0° С до температуры кипения; пл. abА0 — теплота, подводимая к воде в процессе парообразования; пл. сеСВ — теплота, затраченная на перегрев пара.
Учитывая, что количество теплоты в процессе р = const равно разности энтальпий 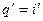 , , площадь, ограниченная ординатами, осью абсцисс и изобарой, проходящей через точку, определяет энтальпию в данной точке. Точка пересечения в верхней и нижней пограничных кривых является критической точкой К.
, , площадь, ограниченная ординатами, осью абсцисс и изобарой, проходящей через точку, определяет энтальпию в данной точке. Точка пересечения в верхней и нижней пограничных кривых является критической точкой К.
Область, лежащая между кривыми аК и сK, — это область влажного насыщенного пара. Область, лежащая правее верхней пограничной кривой, — область перегретого пара.
Исследования паровых процессов и расчеты существенно облегчаются при наличии подробной Т — s-диаграммы, в которой нанесены обе пограничные кривые, сетка изобар и изохор, а также кривые постоянной сухости х = const, которые на рис. 9.8 показаны пунктирными линиями.
§ 6. i—s-диаграмма водяного пара
 Для изучения и расчетов различных термодинамических процессов, в которых рабочим телом является насыщенный и перегретый пар, особо удобна i — s-диаграмма (рис. 9.9).
Для изучения и расчетов различных термодинамических процессов, в которых рабочим телом является насыщенный и перегретый пар, особо удобна i — s-диаграмма (рис. 9.9).
В системе координат i — s наносятся пограничные кривые, изобары и изотермы. Нижняя пограничная кривая и верхняя пограничная кривая строятся по известным значениям 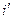 ,
, 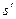 ,
, 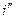 ,
, 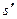 и сливаются в критической точке К. В области влажного насыщенного пара наносятся линии постоянной сухости (пунктирные кривые). В этой диаграмме теплоты жидкостей, парообразование и перегрев изображаются линейными отрезками, а не площадями. Теплота парообразования по данной изобаре
и сливаются в критической точке К. В области влажного насыщенного пара наносятся линии постоянной сухости (пунктирные кривые). В этой диаграмме теплоты жидкостей, парообразование и перегрев изображаются линейными отрезками, а не площадями. Теплота парообразования по данной изобаре
равна разности ординат точек пересечения изобары с правой и левой пограничными кривыми.
Для процесса парообразования, происходящего при р = const,
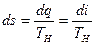 т.е.
т.е. 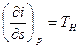
Следовательно, в области влажного насыщенного пара изобары, являясь одновременно и изотермами, представляют собой прямые линии с угловым коэффициентом, равным Tн; из диаграммы видно, что изобары пересекают пограничные кривые без излома. Изохоры, изобары и изотермы в области перегретого пара строятся по точкам. Изобары и изохоры в области перегрева — слабо вогнутые логарифмические кривые; изотермы в области перегретого пара — выпуклые кривые, поднимающиеся слева вверх направо. Вид изотерм определяется температурой, которой они соответствуют. Чем больше температура, тем выше располагается изотерма. Чем дальше от пограничной кривой (х = 1) проходит изотерма, тем больше она приближается к горизонтали i = const, так как в области идеального газа энтальпия однозначно определяется температурой. На рис. 9.9 точки A, В, С изображают соответственно состояния влажного, сухого и перегретого пара. Причем точка А лежит на пересечении изобары (изотермы) и линии постоянной сухости, точка В лежит на пересечении изобары и верхней пограничной кривой, точка С находится на пересечении изобары и изотермы. По положению точки, соответствующей некоторому состоянию пара, можно определить на i — s-диаграмме числовые значения всех параметров в этой точке.
Большинство газов, применяемых в технике, содержит пары тех или иных жидкостей. Наиболее распространенными являются смесь воздуха или какого-либо другого газа с водяным паром, смесь воздуха с парами бензина, керосина и т. п.
Характер изменения параметров парогазовой смеси имеет важное значение в расчетах процесса сушки, кондиционирования воздуха, сверхзвуковых аэродинамических труб, обледенения самолетов, процесса испарения топлива в двигателях и форсировании их впрыском жидкостей и т.д.
Смесь, состоящая из сухого газа и перегретого пара, называется ненасыщенным влажным газом, а смесь из сухого газа и насыщенного пара — насыщенным влажным газом.
При охлаждении влажного газа до определенной температуры (температуры точки росы) пар становится насыщенным, а в дальнейшем может и сконденсироваться.
Состояние парогазовой смеси определяется сравнительно узким диапазоном температуры и давления. Значительное повышение температуры или понижение давления приводит к тому, что влажный газ превращается в простую газовую смесь (гл. 11, § 4).
Полагая, что перегретый пар любой жидкости, входящий в состав влажного газа, приближается по своим свойствам к газам, можно рассматривать влажный газ как газовую смесь.
По закону Дальтона давление смеси идеальных газов р равно сумме парциальных давлений
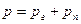 . (9.43)
. (9.43)
где pv — парциальное давление сухого газа; рп — парциальное давление пара.
Равным образом можно записать
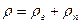 (9.44)
(9.44)
Равенство (9.44) показывает, что плотность влажного газа выше плотности сухого тогда, когда давление влажного газа по уравнению (9.43) выше сухого.
Основными характеристиками влажного состояния газа являются:
относительная влажность j, которая определяет степень насыщения газа паром
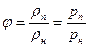 (9.45)
(9.45)
где рп и рн — плотности перегретого и насыщенного пара;
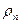 и
и 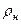 — соответствующие парциальные давления.
— соответствующие парциальные давления.
Соотношение (9.45) справедливо только тогда, когда можно считать, что пар жидкости является идеальным газом вплоть до состояния насыщения. При этом
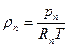 ;
; 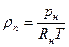 ,
,
где Rп = Rн — газовая постоянная пара;
абсолютная влажность D, определяющая массу пара, содержащегося в 1 м 3 газа,
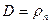 кг/м 3
кг/м 3
влагосодержание d — это масса пара, содержащегося в 1 кг сухого газа,
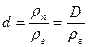
или, определяя рп и рг из уравнения состояния, получим
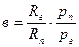 (9.46)
(9.46)
Рассматривая влажный газ как газовую смесь, выведем соотношения, связывающие параметры влажного газа. Пусть состояние, влажного газа определяется его давлением р, температурой t, плотностью r и относительной влажностьюj. По таблицам сухого насыщенного пара определяем для данной температуры значения rн и рн.
Плотность пара в смеси по уравнению (9.45) равна

а плотность сухого газа
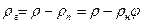 (9.47)
(9.47)
Парциальное давление сухого газа можно определить из уравнения состояния
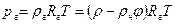
Парциальное давление пара в смеси
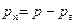
Если заданы для влажного газа р, t,  , а плотность его неизвестна, то, найдя по таблицам насыщенного пара рн и
, а плотность его неизвестна, то, найдя по таблицам насыщенного пара рн и 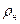 для данной температуры, определим
для данной температуры, определим
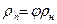
Парциальные давления пара и сухого газа вычислим по формулам
 ,
, 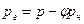
Плотность сухого газа найдем из уравнения состояния
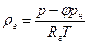 (9.48)
(9.48)
а плотность влажного газа вычислим по формуле (9.44). Влагосодержание на 1 м 3 и на 1 кг сухого газа определяют по формулам:
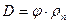 ;
;
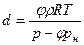 (9.49)
(9.49)
Если газ насыщен паром, то j = 1 и
, а 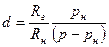 (9.50)
(9.50)
Массовые доли сухого газа и пара во влажном газе соответственно равны:
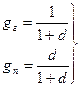 (9.51)
(9.51)
Используя обычное выражение газовой постоянной для смеси газов (гл. 11, § 4), получим
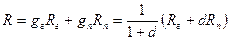 (9.52)
(9.52)
Теплоемкость влажного газа можно определить, зная массовый состав его и теплоемкости сухого газа и пара,
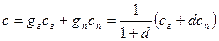 (9.53)
(9.53)
Так же, как и теплоемкость, энтальпия влажного газа равна сумме энтальпий сухого газа и пара. Следовательно,
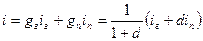 (9.54)
(9.54)
Энтальпия 1 кг сухого газа
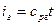
Энтальпия водяного пара, который находится в перегретом состоянии, определяется по формуле
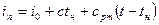 (9.55)
(9.55)
где i0+ctн — энтальпия сухого насыщенного пара в газе (tн— температура кипения при определенном парциальном давлении); срт — средняя теплоемкость перегретого пара.
Для водяного пара iп может быть взята из таблиц водяного пара. Таким образом, энтальпия влажного насыщенного пара равна
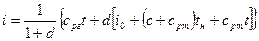 ( 9.56)
( 9.56)
Тепловые процессы парогазовой смеси имеют ряд особенностей, их можно разделить на:
процессы, идущие без фазовых превращений, в этом случае относительная влажность j р2 > р3 и т. д.), изохоры
Дата добавления: 2015-04-03 ; просмотров: 3361 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
http://zaochnik.com/spravochnik/fizika/molekuljarno-kineticheskaja-teorija/uravnenie-sostojanija-veschestva/
http://helpiks.org/3-2548.html