Стехиометрические и кинетические уравнения химических реакций
Математическое описание химических реакций .
Самый сложный класс химико-технологических процессов – это процессы протекания химических реакций (реакционные процессы). Именно к ним наиболее важно применять методы математического моделирования. В первую очередь мы будем рассматривать моделирование реакционных процессов. Математическое описание складывается из описания стехиометрии, химического равновесия и кинетики .
Стехиометрия и равновесие химических реакций
Стехиометрическое уравнение химической реакции (обычно его называют просто уравнением реакции) – это, как правило, самая первая информация, получаемая о ней химиком. Стехиометрическое уравнение представляет собой краткое выражение материального баланса реакции. Например, уравнение
означает, что всякий раз, как в процессе реакции затрачиваются две молекулы Н 2 , одновременно расходуется ровно одна молекула О 2 и образуются две молекулы H20
Наряду с подобными записями «со стрелкой», часто пишут уравнения реакций со знаком равенства. Мы будем считать, что оба способа записи равноценны, и в разных случаях пользоваться тем из них, который оказывается удобнее .
В большинстве случаев реагирующие вещества будем обозначать не конкретными формулами, а в общем виде : А, В, С, . J . причем под символом J обычно будем понимать «какое-либо вещество»; то, что относится к J, можно отнести к любому из веществ . Иногда все вещества будем обозначать одной буквой А , различая индексами : A 1 , A 2 и т. д. Стехиометрические коэффициенты в общем виде обозначим s с индексом вещества : s a , s j .
В расчетах сложных реакций бывают случаи , когда уравнения целесообразно писать не только в привычном виде
или в общем виде
причем в двух последних уравнениях разница между исходными веществами (реагентами) и продуктами реакции отражена в различных знаках стехиометрических коэффициентов . Обычно при таком способе записи считают, что для реагента или исходного вещества (расходуемого в реакции) s а для продукта (образующегося вещества ) s >0. Если реакция состоит из ряда стадий, то получается система из п уравнений :
где ( i – номер вещества ; m – общее количество веществ; п – число стадий. Разумеется, часть стехиометрических коэффициентов в любой строке может быть равна нулю .
Стехиометрические балансы . Стехиометрические расчеты особенно просты, когда количество вещества выражается в молях . Всюду, кроме особо оговоренных мест, мы будем выражать количество вещества g в молях, а концентрацию с – в молях на литр (или, что то же самое, в киломолях на кубометр ).
Рассмотрим реакцию, аналогичную выражению ( 1 )
Индексом 0 будем обозначать начальный момент ( c jo — начальное количество вещества J ).
Из уравнения реакции (3) для любого момента реакции вытекают уравнения стехиометрического баланса
Уравнение (4) выражает баланс реакции (1) по водороду: слева – удвоенное количество грамм-атомов Н в системе в текущий момент, справа – равное ему количество в момент начальный .
Уравнение (5) – баланс по кислороду. Первый член слева – количество грамм-атомов О в молекулах О 2 (поскольку в каждой молекуле содержится 2 атома ).
Левая и правая части уравнения (6) равны избытку одного из реагентов сверх стехиометрии . Если в избытке будет O 2 , то разность положительна, если Н 2 , то отрицательна. Если избытка нет, разность равна нулю. В ходе реакции избыток не меняется .
Если реакция идет при постоянном объеме (изохорически), то в любых балансовых уравнениях можно заменить g на с. Там, где не сделано оговорок, будем считать реакцию изохорической .
Уравнение стехиометрического баланса – одно из выражений стехиометрической эквивалентности реагирующих веществ. В реакции (3) эквивалентность определяется соотношениями :
В соотношениях эквивалентности для одностадийных реакций коэффициенты равны (или пропорциональны) стехиометрическим коэффициентам и обратны коэффициентам уравнений баланса (4) – (6 ).
Левые части уравнений (4) – (6) обладают тем свойством, что они не меняются по ходу реакции . Поэтому их называют и н-вариантами реакции ( invariantus – по-латыни неизменный), а для многостадийных реакций – инвариантами системы реакций .
Уравнения стехиометрического баланса (или инварианты) позволяют решать ряд важных расчетных задач . При описании химических процессов одна из важнейших моделей – уравнение материального баланса (3.5) или (3.6 ). Неизвестные, входящие в эти уравнения, – либо количества, либо концентрации веществ . Конкретный вид систем уравнений (3.5) может быть весьма сложным, и для упрощения вычислений часто крайне желательно уменьшить число уравнений, а стало быть, уменьшить число неизвестных g J . Для этого и могут служить стехиометрические балансы .
Так , из уравнений (4) – (6) следуют равенства
позволяющие исключить из дальнейших вычислений две величины ( g В и g С ), выразив их через g А и начальные условия .
Аналогично решается и другая задача: расчет состава реакционных смесей при неполном задании концентраций. Зная концентрации части веществ и все исходные концентрации, удается рассчитать концентрации других веществ, не измеряя их; иногда невязка стехиометрического баланса сигнализирует либо об ошибках в определении концентраций, либо о неверно записанной схеме реакций (например, о неучтенной побочной реакции ).
При решении подобных задач не следует учитывать обратимость реакций: стехиометрические балансы не зависят от обратимости .
Рассмотрим задачу. При постоянном объеме проходит сложная реакция :
В начальный момент концентрации составляли : с А0 = 5 моль/л ; с В0 = 6 моль/л ; С c0 =С 0 = C do =C fo =C ho =0.
В некоторый момент t смесь проанализировали, определив концентрации : с а =1 моль / л ; c С =2 моль/л ; c Е =2 моль/л ; c Н =5 моль/л . Требуется рассчитать концентрации В , D и F .
Решая эту задачу, необходимо иметь в виду, что все стадии сложной реакции идут в реакционной смеси одновременно .
Наиболее общий метод решения стехиометрических задач, использующий аппарат линейной алгебры, будет изложен ниже. Сейчас познакомимся с простым способом решения, связанным с представлением материального баланса реакции в виде ориентированного графа .
Граф – это система точек (вершин), соединенных линиями (ребрами). Если на каждом ребре указано направление (ребро – это стрелка), граф называют ориентированным. На рис. 1 изображен граф материального баланса данной реакций из расчета на 1 литр .
Вершины графа означают : 1– исходное количество А; 2 – количество А, оставшееся к моменту t ; 3 – израсходованное количество А; 4 – доля израсходованного А, пошедшая на образование С; 5 – соответствующая доля, пошедшая на образование D; 6 – исходное количество В; 7 –количество В, оставшееся к моменту t ; 8 – израсходованное количество В; 9 – количество образовавшегося С; 10 – количество С оставшееся к моменту C ; 11 – расход С на образование Е и Н (обратите внимание на то, что Е и Н образуются из одних и тех же молекул С, поэтому отдельно на образование Е и на образование Н вещество С не расходуется); 12 – количество образовавшегося Е, целиком оставшееся к моменту t , поскольку Е в дальнейшие реакции не вступает; 13 – количество Н, образовавшегося из С; 14 – количество образовавшегося D; 15 – количество D, оставшееся к моменту t ; 16 – израсходованное количество D; 17 – количество образовавшегося F ; 18 – количество Н, образовавшегося из D ; 19 – общее количество образовавшегося Н .
Расчет проводится от тех вершин, для которых количества известны, к другим вершинам. В начале известны количества А в вершинах 1 и 2 (5 и 1 моль). Отсюда количество А в вершине 3 равно 4 молям .
Дальнейший расчет ведем от вершины 12. Поскольку из С образовалось 2 моля Е, расход С составил также 2 моля (вершина 11). Вместе с 2 молями оставшегося С (вершина 10) получаем в вершине? количество образовавшегося С, составляющее 4 моля. Теперь переходим из вершины 11 в вершину 13; при расходовании 2 молей С, наряду с веществом Е, образуется 2 моля Н .
Следующий этап – расчет количеств А и В, пошедших на образование 4 молей С . По стехиометрии на это затрачено 2 моля А (вершина 4) и 4 моля В (вершина 8) . Следовательно, количество В, оставшегося к моменту t , равно 6 – 4=2 молям (вершина 7) .
Переходим к расчету образования и превращения D. Из количеств в вершинах 3 и 4 следует, что на образование D затрачено 2 моля А (вершина 5) . По стехиометрии из них образовалось 4 моля D – вершина 14 . Далее расчет приходится вести от вершины 18 . Поскольку общее количество образовавшегося Н равно 5 молям (вершина 19), а из С образовалось 2 моля Н (вершина 13), то в вершине 18 имеем 3 моля образовавшегося Н. На эту реакцию израсходовано 3 моля D (вершина 16). Одновременно по стехиометрии образовалось 6 молей F (вершина 17) . Наконец, сопоставив вершины 14 и 16, найдем, что из 4 молей образовавшегося D не вступил в реакцию и остался 1 моль (вершина 15) .
Окончательно : с В =2 моль/л; с d =1 моль/л ; с F =6 моль/л .
Применение линейной алгебры для решения стехиометрических задач .
Для многостадийных реакций нахождение инвариантов удобно производить методами линейной алгебры .
Условно рассмотрим выражение (2) как систему из п уравнений с т неизвестными A j . Пусть b 1 , b 2 , . , b т — какое-либо решение этой системы, т. е. при подстановке в систему (2) вместо A i чисел hi она обращается в тождество . Тогда сумма
является инвариантом данной системы реакций .
Уравнения системы (2) однородны (правые части их равны нулю) . При этом всегда m rg , где rg – ранг матрицы системы (2) . Такая система имеет бесконечное число решений . Поэтому для любой реакции можно определить бесконечное число инвариантов. Но только часть из них , в количестве, равном т – rg , линейно независимы. Любой другой инвариант может быть получен как линейна я комбинация выбранных ( т – rg ) инвариантов, соответствующих фундаментальной системе решений уравнений (2) .
Напомним , что линейной комбинацией величин или выражений x 1 , x 2 , . , x k называется выражение l 1 x 1 + l 2 x 2 +. + l k x k , где l 1 , . l k – произвольные числа . Если в некоторой системе выражений ни одно из них не может быть представлено как линейная комбинация остальных , система называется линейно независимой . Частный случай линейной комбинации – сумма ( l 1 = l 2 = ? = l k =1). Подробно об однородных системах, линейно независимых решениях, ранге и фундаментальных системах решений рассказано в книге [17]. Мы лишь поясним сказанное примерами .
Реакции А ® В соответствует уравнение
для которого т= 2, rg = l . Фундаментальная система состоит из 2–1=1 решения – например, b 1 = l , b 2 ,=1, которому соответствует инвариант g \+ gB , в данном простейшем случае линейная комбинация получается единственным образом – умножением фундаментального решения на произвольное число . Действительно, выражения 2 g A + 2 g B ; 750 g A + 750 g B ; 0,112 g A + 0,112 g B и т. д. также, разумеется, будут инвариантами этой реакции .
Для уравнения , соответствующего реакции (3) , m =3 , rg = l . Здесь фундаментальная система состоит из двух решений . Если в качестве одного из них принять : b 1 = l , b 2 =0, b 3 =1, а в качестве второго b 1 =0, b 2 =2, b 3 =1, то получим инварианты , определяемые , балансами (4) и (5). Любая линейная комбинация обоих решений также явится решением , и ей будет соответствовать инвариант реакции (3) . Так, вычтя второе решение из первого, получим b 1 =+ l , b 2 = –2 , b 3 =0 ; соответствующий инвариант g A –2 g B дан уравнением (6) .
Пример 1 . Инварианты сложной реакции .
Рассмотрим процесс нитрования бензола . При этом пренебрежем образованием о – и n –динитробензолов и несимметричных тринитробензолов. Тогда схема реакции запишется так :
Обозначим для краткости С 6 Н 6 =А 1 ; C 6 H 5 NO 2 = A 2 ; C 6 H 4 ( NO 2 ) 2 = A 3 ; С 6 Н 3 ( NO 2 ) 3 =А 4 ; Н 2 О=А 5 ; Н N О 3 =А 6 ; NO 2 =А 7 ; O 2 = A 8 ; N 2 O 4 =А 9 . Однородная система уравнений может быть записана в виде :
Методом Гаусса систему (8) привели к треугольному виду
Если принять, например, в качестве свободных переменных А 3 , А 4 , A 8 и А 9 , то получается фундаментальная система из четырех решений :
которой соответствуют инварианты :
Линейно независимые стадии реакции .
Рассмотрим для примера синтез карбамида. Можно предполагать следующие его стадии :
а также ряда других, не приводимых здесь для простоты ( например , образование NH 4 OH свободных радикалов и т. д.). Задача физико-химического изучения реакции заключается в расшифровке механизм а, т. е. выявлении того, какие из стадий проходят в действительности и каковы их скорости. Но некоторые практические задачи могут быть решены и без такой расшифровки. Так, равновесие и тепловой эффект не зависят от того, через какие стадии проходит реакция. Не нужна расшифровка механизма и для определения такого важного технологического показателя, как выход карбамида .
При решении этих задач оказывается, что записанная система стехиометрических уравнений в некотором смысле избыточна . Действительно, если мы исключим из рассмотрения стадию ( в ), то на ходе и результатах расчета равновесия или выхода это практически не отразится , поскольку данная стадия получается как сумма стадий ( а ) и ( б ) . То же относится к стадии ( д ), равной сумме ( а ) + ( г ).
В любой обратимой реакции одна из стадий равна другой, умноженной на – 1 . Некоторые стадии могут представлять собой не просто суммы, но и более сложные линейные комбинации иных стадий: так, ( и )=( б ) – ( г ) – ( а ) .
Разумеется, при исследовании механизма можно исключить из рассмотрения лишь такие стадии, для которых установлена нулевая скорость. Но в вышеназванных ограниченных задачах часто целесообразно исключить те стадии, которые можно получить как линейную комбинацию других стадий .
Сведение системы типа (2) к линейно независимой путем исключения строк, являющихся линейными комбинациями других, часто упрощает расчеты. Например, равновесие в системе характеризуется таким числом констант равновесия, которое равно числу линейно независимых строк. Остальные константы могут быть рассчитаны, исходя из предыдущих. Исключение линейно зависимых строк не меняет инвариантов данной системы реакций .
Для определения числа линейно независимых стадий записывают систему уравнений (2) для данной реакции. При этом, в соответствии со сказанным выше, из каждой обратимой реакции нужно включать в систему лишь одну стадию (безразлично, какую). Далее выписывают матрицу коэффициентов этой системы
Число линейно независимых стадий равно рангу матрицы . На вопрос о том, какие именно стадии включить в систему линейно независимых, можно дать не один ответ: любая совокупность из k строк матрицы , содержащая хотя бы один минор k- го порядка, не равный нулю, линейно независима. В таких случаях выбор требуемой системы определяется соображениями удобства – например, тем, для каких именно стадий известны константы равновесия. Более того, иногда удобно включить в линейно независимую систему новые стадии, полученные как линейные комбинации части строк системы (3), и исключить часть последних.
Способы нахождения ранга матрицы и линейно независимых комбинаций строк рассматриваются в линейной алгебре; когда число строк достаточно велико, целесообразно использовать ЭВМ .
ПРИМЕР 2 . Расчет линейно независимых стадий .
Для реакции синтеза карбамида обозначим : CO 2 = A 1 ; N Н 3 =А 2 ; Н 2 О=А 3 ;
NH 2 СОО N Н 4 =А 4 ; N Н 2 СО N Н 2 =А 5 ; ( N Н 4 )СО 3 =А 6 ; N Н 4 НСО 3 =А 7 . Тогда система ( 2 ) примет вид :
Запишем матрицу для этой системы:
Было рассчитано, что ранг матрицы , rg =4. Таким образом, из всех стадий реакций в качестве линейно независимых могут быть приняты только 4. Например, это могут быть стадии ( а ), ( б ), ( г ), ( е ) . Проверка показывает, что для этих стадий rg =4, и они действительно линейно независимы .
Подробно вопросы стехиометрических расчетов для сложных реакций рассмотрены в книге [17] .
Степень превращения и селективность – две характеристики технологического процесса, расчет которых тесно связан со стехиометрическими соотношениями .
Степень превращения х какого-либо реагента А равна доле превратившегося в продукты вещества от общего начального количества этого вещества. Например, для реакции :
степень превращения вещества А можно выразить следующим образом:
а степень превращения вещества В так :
Величины х а и х В в общем случае не равны одна другой . Они равны только, если соблюдается соотношение
т. е. если А и В взяты в стехиометрических количествах. В противном случае для того из веществ, которое взято в избытке относительно стехиометрии, степень превращения окажется меньше. Если избыток велик, вещество будет израсходовано лишь в малой степени, тогда как для второго степень превращения может быть близкой к 1. Этим нередко пользуются в технологии: давая в избытке менее ценный реагент, сдвигают равновесие и приближают использование ценного исходного вещества к 100% .
При постоянном объеме в формуле ( 10 ) можно заменить g на с .
Расчет степени превращения можно проводить и через количество продукта . Так, в реакции
степень превращения А составит
Но в такой форме расчет менее удобен, особенно если реакция—многостадийная. Так же, как описанный ниже расчет селективности, расчет степени превращения удобнее вести через исходное вещество .
Отметим еще следующее . Количество реагента можно выразить формулой
В этом смысле с точностью до знака степень превращения можно рассматривать как безразмерную концентрацию. Во многих работах описание протекания химических реакций проводится именно в терминах степеней превращения .
Селективность , под которой понимают выход целевого продукта на затраченное исходное вещество, является одной из важнейших характеристик тех процессов, в которых наряду с основной реакцией протекают побочные стадии, приводящие к образованию нежелательных побочных продуктов . Селективность s может быть определена следующим образом :
Таким образом , величина s показывает, какая доля превратившегося исходного вещества затрачена на основную реакцию—на образование целевого продукта .
При расчете селективности часто необходимо учитывать стехиометрическую эквивалентность исходных веществ и продуктов. Например, проходит реакция, в которой В – целевое вещество, а остальные продукты – отбросы .
В реакционной смеси : с А = 0,2 моль/л ; с В = 2,6 моль/л ; с С = 0,6 моль/л ; с D = 0,4 моль/л ; с Е = 0,2 моль/л ; с Н = 1,5 моль/л . Требуется рассчитать s .
Установим стехиометрическую эквивалентность веществ :
Теперь легко получить
В числителе – количество А, затраченное на образование В; второе и третье слагаемые знаменателя – затраты А на образование С и Е . Затраты А на получение Н не учитываются, поскольку образование Н не является самостоятельной , побочной реакцией: этот продукт обязательно образуется при получении В; при этом А отдельно не расходуется . По этой же причине в выражении для s учитывается образование лишь одного из продуктов второй стадии (безразлично, которого – для примера в уравнение введено вещество С) .
Расчет химического равнове сия основывается на решении систем уравнений , задаваемых константами равновесия , совместно с уравнениями стехиометрического баланса , аналогичными уравнениями ( 4) – (6 ). При этом в случае многостадийных реакций следует рассматривать только линейно независимые стадии , иначе система уравнений окажется избыточной , что может привести , например , к вырождению матрицы коэффициентов .
Мы рассмотрим лишь простейший случай , когда константы равновесия достаточно точно выражаются через концентрации . Трудности , связанные с введением коэффициентов активности , обычно разбираются в курсе физической химии . Но и без этих трудностей расчет равновесия далеко не всегда тривиально прост .
Пример 3 . Расчет равновесия .
Рассчитаем равновесные концентрации веществ в реакции
если заданы константы равновесия всех стадий, а также известно, что в исходной смеси концентрация реагента составляла , а остальные вещества отсутствовали.
Задача сводится к решению системы
Из первых трех уравнений легко получить
Тогда четвертое уравнение получит вид :
Дальнейший расчет элементарен .
Если в прямой или обратной реакции какой-либо стадии участвуют более одной молекулы, система оказывается нелинейной, что усложняет решение .
Формальная химическая кинетика
Кинетика химических реакций – основа их описания. Главной проблемой кинетики как раздела физической химии является вопрос о механизме реакции. Основная задача кинетики – раскрыть механизм реакции и установить, как он отражается в кинетических закономерностях .
При анализе, описании и расчете протекания реакции как элемента химико-технологического процесса вопрос о механизме реакции часто не встает: кинетические закономерности рассматриваются как уже заданные (например, изученные на предыдущем – физико-химическом этапе исследования). В этих случаях описание проводится на языке формальной кинетики .
Скорость реакции – основное понятие кинетики . Она определяется как количество вещества , реагирующее в единицу времени в единице реакционного пространства
где V – реакционное пространство , которое в случае гомогенной реакции представляет собой объем , а в случае гетерогенной – поверхность .
Часто применяют удобный частный вид уравнения (1). Но прежде, чем записать его, необходимо подчеркнуть, что пользоваться им можно только при соблюдении двух условий :
1 ) реакция проходит при постоянном объеме ;
2) объем этот можно считать закрытым .
Закрытой, или замкнутой называется система, которая по ходу процесса не обменивается веществом с окружающей средой (хотя и может обмениваться энергией). Большинство химико-технологических процессов, и в первую очередь все непрерывные процессы, протекают в открытых системах, которые характеризуют обмен с окружающей средой энергией и веществом. Лишь в одном крайнем случае (поток идеального вытеснения) в аппарате непрерывного действия удается выделить объем , который можно считать закрытым ( см . раздел 12 ).
Если гомогенная реакция в закрытом объеме проходит изохорически ( V = const ), то постоянную величину 1 / V в формуле (1) можно внести под знак дифференциала ; учтя , что g / V = c есть концентрация, получим —
Уравнением (2) часто пользуются , так как оно удобно для интегрирования ; но необходимо помнить , что это – лишь частный случай , который верен далеко не всегда .
Формально простые и сложные реакции . Химическая кинетика определяет простые (одностадийные) реакции как такие, которые по существу проходят в одну стадию; простая реакция содержит один элементарный акт. Однако такие реакции, которые проходят как истинно одностадийные, встречаются редко .
В формальной кинетике оказывается удобным говорить о формально простых реакциях. Так называют реакции, которые можно формально представить как протекающие в одну стадию. При этом по существу реакция может быть сложной, проходящей через какие-то промежуточные стадии. Но если в условиях рассматриваемой задачи промежуточные продукты не обнаруживаются, то реакция будет считаться формально простой. В какой-либо другой задаче та же реакция может фигурировать, как сложная – это будет означать лишь переход к иному формальному рассмотрению. Несмотря на условность понятия – формально простая реакция, оно удобно и поэтому часто используется .
Многие реакции всегда приходится рассматривать как сложные: они явно распадаются на стадии (продукты различных стадий образуются в значительных количествах) .
Можно выделить три простейших типа сложных реакций .
Обратимая реакция . С излагаемой точки зрения это сложная реакция, состоящая из двух стадий: прямой и обратной реакции .
Параллельные реакции : исходное вещество по двум или нескольким параллельно протекающим реакциям (стадиям) превращается в два или несколько продуктов. Примером может служить нитрование толуола с параллельным получением о- и n — нитротолуолов .
Последовательные реакции : стадии реакции следуют одна за другой, продукт первой стадии является исходным веществом второй, и т. д. Примером может служить деполимеризация с последовательным распадом макромолекулы на все более мелкие части. Остальные сложные реакции можно представить в виде комбинаций указанных трех типов – например, последовательно-параллельные, последовательные реакции с обратимыми стадиями
Оговоримся, что здесь мы не рассматриваем такие многостадийные реакции, стадии которых разделены во времени: сначала проводится одна стадия, затем через некоторое время – вторая и т. д. Такой процесс мы будем считать несколькими независимыми реакциями. А те сложные реакции, о которых здесь идет речь, проходят так, что в реакционной смеси одновременно идут все стадии .
Скорость одностадийной реакции и скорость реакции по веществу .
Рассмотрим формально одностадийную реакцию (она может быть и одной из стадий сложной реакции)
Возникает следующий вопрос. Количество какого из веществ (А, В или С) следует вводить в уравнение (1) при определении скорости реакции (3)? Ответ на этот вопрос легко получить из понятия стехиометрической эквивалентности. Если за какой-то промежуток времени прореагирует некоторое количество g A вещества А, то за это же время количества прореагировавшего В и образовавшегося С составят :
Поэтому , если мы подставим в формулу (1) один раз g а , другой раз § в , третий раз g c , то получим три значения : r A , r B и r C , отличающиеся одно от другого постоянными множителями . Строго говоря , это одна и та же величина – скорость реакции (3), но выражена она в разных единицах . Для единообразия скорость одностадийной реакции , или скорость стадии принято определять , деля скорость , выраженную через любое вещество J , участвующее в этой стадии , на стехиометрический коэффициент этого вещества
В этом случае безразлично, какое из веществ принято в качестве J. Действительно, из выражений (4) следует :
Во многих формально-кинетических расчетах используются и значения скорости стадии, выраженные через конкретные вещества. Для этих значений из формулы (5) получаем :
При пользовании формулой (7) следует учитывать, что если в рассматриваемой стадии J – исходное вещество , то s j и соответственно, получим r J J – продукт, т. е. образуется на данной стадии , то s j >0 и r J >0. Так, в реакции (3) s А s b s С >0, и соответственно , r а r В r С >0 .
Пример 1 . Скорость реакции по веществам .
имеющую первый порядок по реагенту А и нулевой – по В ; константу скорости обозначим через k . Для этой реакции, в соответствии с формулой (7), имеем
r а = – 2 kc A r B = – kc A r C = 2 kc A
Обратите внимание на то, что и r а , и r B и r C зависят от c A и не зависят от c B . Вопрос о том, почему это так, лежит вне формальной кинетики – это вопрос о механизме реакции. Но в формально-кинетических расчетах важно помнить, что в случае формально одностадийной реакции или стадии сложной реакции r а , r B и r C – не три разные скорости, а одна и та же скорость реакции, и различаться эти величины могут только множителями – стехиометрическими коэффициентами. Уравнение реакции устанавливает однозначную связь между реагирующими количествами А, В и С и, стало быть, связь между скоростями по этим веществам, определяемую формулой (7) .
Порядок реакции . Скорость многих (хотя и не всех) формально простых реакций, а также стадий сложных реакций пропорциональна концентрациям реагирующих веществ в некоторых степенях. Показатели степени в таком случае называют порядком реакции по реагентам. Так, для реакции
соответствующая зависимость будет выражаться кинетически м уравнением
Здесь n 1 — порядок реакции по веществу А , n 2 – порядок по веществу В , сумму n 1 + n 2 называют общим, или суммарным порядком. Коэффициент пропорциональности k именуют константой скорости реакции .
Порядок реакции или стадии будем обозначать цифрой над (или под) стрелкой, указывающей направление реакции; здесь же будем приводить обозначение константы скорости . Так, запись
обозначает обратимую реакцию, у которой прямая стадия имеет 1-й порядок по А и константу скорости – k 1 , обратная – 2-й порядок по С и константу k 2 . Поскольку порядок по В не указан, он – нулевой .
На практике встречаются реакции самых разнообразных порядков: целочисленных и дробных, нулевого, изредка и отрицательных. Численное значение порядка может быть и очень малым, и большим – так, для каталитической димеризации ацетилена установлен порядок выше 9-го [50] .
Достаточно часты случаи, когда порядок по каждому из реагентов совпадает с его стехиометрическим коэффициентом. Но обязательно такое совпадение лишь для строго (не формально) простых реакций. Вследствие сложности механизма формально одностадийные процессы могут протекать по порядкам, сильно отличным от стехиометрических коэффициентов .
Заметим, что у тех реакций, порядок которых не совпадает со стехиометрическими коэффициентами, при приближении к равновесию (или к 100%-ному. Превращению, если реакция необратима), как правило, начинается изменение порядка – непосредственно вблизи равновесия порядок сходится к величине, определяемой стехиометрией .
Существует достаточно много реакций, скорость которых вообще не может быть описана формулой (9). Понятие – порядок реакции к таким реакциям либо вообще неприменимо, либо применимо с оговорками. В частности, обычно нельзя говорить о порядке разветвленных цепных реакций. Для многих неразветвленных цепных реакций и гетерогенно-каталитических реакций (в случае существенного влияния стадии адсорбции) в кинетическое уравнение, наряду со степенными, входят и сомножители более сложной структуры. Например, кинетическое уравнение для синтеза бромистого водорода из простых веществ имеет вид :
В то же время задание порядка реакции иногда является кратким выражением особенностей кинетики .
Пример 2. Гомогенный катализ. Реакция может иметь некоторый порядок по веществу, не входящему в стехиометрическое уравнение :
Это гомогенный катализ: вещество D является катализатором. В роли гомогенного катализатора может выступать один из продуктов реакции — это случай автокатализа :
По некоторым веществам порядок реакции может быть отрицательным – отрицательный катализ, или ингибирование. Чаще всего в роли ингибитора выступает один из продуктов реакции :
Хотя возможен и случай, когда ингибитор не отражен в стехиометрии реакции :
Температурная зависимость скорости реакции . В уравнении (9) концентрации не зависят от температуры в явном виде. Порядок реакции иногда претерпевает изменения с ростом температуры, но это – скорее исключение, чем правило. Наиболее тесно с температурой связана константа скорости реакции. Эта зависимость чаще всего может быть описана уравнением Аррениуса
Параметрами уравнения (12) являются предэкспоненциальный множитель , или предэкспонента А и энергия активации E . Формальная кинетика не занимается физическим смыслом величин А и Е, но для многих задач важно понимать, какая именно характеристика реакции определяется величиной Е .
Иногда приходится встречаться с мнением, что чем больше энергия активации, тем, в соответствии с формулой (12), меньше величина k и значит, тем медленнее реакция. Но это мнение неверно. Величина k определяется не значением Е , а совокупностью значений Е и А. Поэтому сама по себе величина Е еще не определяет, быстра или медленна реакция .
Формальный (феноменологический) смысл Е иной: чем больше энергия активации, тем сильнее скорость реакции зависит от температуры. На рис. 1 изображена зависимость констант скоростей двух одностадийных реакций от температуры. По рисунку сразу можно сказать, что Е 2 > Е 1 .
Указанный характер влияния Е на ход реакции позволяет произвести быструю качественную оценку влияния температуры на ход некоторых сложных реакций. Так, для обратимых реакций получаются следующие закономерности .
Обратимые экзотермические реакции характеризуются соотношением E 2 > E 1 : энергия активации обратной реакции больше, чем прямой. Поэтому с ростом температуры k 2 растет быстрее , чем k 1 , в результате чего равновесие смещается влево. Наоборот, в эндотермических обратимых реакциях E 2 E 1 , и с ростом температуры равновесие смещается вправо .
Рис . 1 . График температурной зависимости констант скорости двух реакций ( E 2 E 1 ).
Если рассматривается сложная реакция, некоторые стадии которой являются для нас побочными, то соотношение энергий активации различных стадий определяет влияние температуры на селективность: при нагревании преимущественно ускоряются те стадии, которые характеризуются большей энергией активности .
Скорость сложных реакций . Для всех типов формально сложных реакций, кроме простейшей обратимой, понятие общей скорости реакции не имеет смысла. Реакция состоит из ряда стадий, каждая из них имеет свою скорость, и нельзя достаточно естественным образом определить, что такое скорость всей сложной реакции в целом .
В то же время любое из участвующих в реакции веществ образуется или расходуется с определенной скоростью, причем знание этой скорости обязательно при расчете процесса .
Скорость сложной реакции по любому веществу J равна алгебраической сумме скоростей всех стадий по этому веществу (с учетом стехиометрических коэффициентов )
Здесь i – номер стадии; m – число стадий .
Скорость стадии по веществу определяется формулой (7). Если вещество в стехиометрии стадии не участвует, его стехиометрический коэффициент в этой стадии равен нулю .
Пример 3 . Скорости сложной реакции по веществам .
Запишем кинетические уравнения для веществ, участвующих в реакции :
Вещество А участвует в 1-й, 2-й и 3-й стадиях со стехиометрическими коэффициентами – 1, +1 и – 1 . Поэтому
( если выражение не ясно, вернитесь к примеру 1 ). Соответственно
Последнее равенство естественно, поскольку Н, будучи катализатором, не участвует в стехиометрии реакций: не расходуется и не образуется .
Заметьте, что если вещество участвует в обратимой реакции, то стехиометрические коэффициенты для прямой и обратной стадий равны по абсолютной величине и обратны по знаку .
Интегрирование уравнений кинетики . Если химическая реакция (простая или сложная) проходит в замкнутой системе при постоянном объеме, то ход ее, как правило, описывается системой дифференциальных уравнений. Действительно, совместное рассмотрение уравнений (2), (9) и (13) приводит к формуле :
причем число таких уравнений равно числу веществ, участвующих в реакции (реагентов и продуктов). Однако всегда можно уменьшить число уравнений, исключив из рассмотрения часть веществ при помощи стехиометрических инвариантов .
Пример 4 . Упрощение системы кинетических уравнений .
Рассмотрим две задачи на описание кинетики . 1 по А) А
Здесь система уравнений (14) имеет вид :
Но простейшие стехиометрические соотношения ( при с В0 = с С0 =0)
Химическая кинетика. Скорость химических реакций
Темы кодификатора ЕГЭ: Скорость реакции. Ее зависимость от разных факторов.
Скорость химической реакции показывает, как быстро происходит та или иная реакция. Взаимодействие происходит при столкновении частиц в пространстве. При этом реакция происходит не при каждом столкновении, а только когда частица обладают соответствующей энергией.
Скорость реакции – количество элементарных соударений взаимодействующих частиц, заканчивающихся химическим превращением, за единицу времени.
Определение скорости химической реакции связано с условиями ее проведения. Если реакция гомогенная – т.е. продукты и реагенты находятся в одной фазе – то скорость химической реакции определяется, как изменение концентрации вещества в единицу времени:
υ = ΔC / Δt
Если реагенты, или продукты находятся в разных фазах, и столкновение частиц происходит только на границе раздела фаз, то реакция называется гетерогенной, и скорость ее определяется изменением количества вещества в единицу времени на единицу реакционной поверхности:
υ = Δν / (S·Δt)
Факторы, влияющие на скорость химической реакции
1. Температура
Самый простой способ изменить скорость реакции – изменить температуру . Как вам, должно быть, известно из курса физики, температура – это мера средней кинетической энергии движения частиц вещества. Если мы повышаем температуру, то частицы любого вещества начинают двигаться быстрее, а следовательно, сталкиваться чаще.
Однако при повышении температуры скорость химических реакций увеличивается в основном благодаря тому, что увеличивается число эффективных соударений. При повышении температуры резко увеличивается число активных частиц, которые могут преодолеть энергетический барьер реакции. Если понижаем температуру – частицы начинают двигаться медленнее, число активных частиц уменьшается, и количество эффективных соударений в секунду уменьшается. Таким образом, при повышении температуры скорость химической реакции повышается, а при понижении температуры — уменьшается .
Обратите внимание! Это правило работает одинаково для всех химических реакций (в том числе для экзотермических и эндотермических). Скорость реакции не зависит от теплового эффекта. Скорость экзотермических реакций при повышении температуры возрастает, а при понижении температуры – уменьшается. Скорость эндотермических реакций также возрастает при повышении температуры, и уменьшается при понижении температуры.
Более того, еще в XIX веке голландский физик Вант-Гофф экспериментально установил, что скорость большинства реакций примерно одинаково изменяется (примерно в 2-4 раза) при изменении температуры на 10 о С.
Правило Вант-Гоффа звучит так: повышение температуры на 10 о С приводит к увеличению скорости химической реакции в 2-4 раза (эту величину называют температурный коэффициент скорости химической реакции γ).
Точное значение температурного коэффициента определяется для каждой реакции.

здесь v2 — скорость реакции при температуре T2,
v1 — скорость реакции при температуре T1,
γ — температурный коэффициент скорости реакции, коэффициент Вант-Гоффа.
В некоторых ситуациях повысить скорость реакции с помощью температуры не всегда удается, т.к. некоторые вещества разлагаются при повышении температуры, некоторые вещества или растворители испаряются при повышенной температуре, т.е. нарушаются условия проведения процесса.
2. Концентрация
Также изменить число эффективных соударений можно, изменив концентрацию реагирующих веществ . Понятие концентрации, как правило, используется для газов и жидкостей, т.к. в газах и жидкостях частицы быстро двигаются и активно перемешиваются. Чем больше концентрация реагирующих веществ (жидкостей, газов), тем больше число эффективных соударений, и тем выше скорость химической реакции.
На основании большого числа экспериментов в 1867 году в работах норвежских ученых П. Гульденберга и П. Вааге и, независимо от них, в 1865 году русским ученым Н.И. Бекетовым был выведен основной закон химической кинетики, устанавливающий зависимость скорости химической реакции от концентрации реагирующих веществ:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ в степенях, равных их коэффициентам в уравнении химической реакции.
Для химической реакции вида: aA + bB = cC + dD закон действующих масс записывается так:

здесь v — скорость химической реакции,
CA и CB — концентрации веществ А и В, соответственно, моль/л
k – коэффициент пропорциональности, константа скорости реакции.
Например , для реакции образования аммиака:
закон действующих масс выглядит так:

Константа скорости реакции k показывает, с какой скоростью будут реагировать вещества, если их концентрации равны 1 моль/л, или их произведение равно 1. Константа скорости химической реакции зависит от температуры и не зависит от концентрации реагирующих веществ.
В законе действующих масс не учитываются концентрации твердых веществ, т.к. они реагируют, как правило, на поверхности, и количество реагирующих частиц на единицу поверхности при этом не меняется.
В большинстве случаев химическая реакция состоит из нескольких простых этапов, в таком случае уравнение химической реакции показывает лишь суммарное или итоговое уравнение происходящих процессов. При этом скорость химической реакции сложным образом зависит (или не зависит) от концентрации реагирующих веществ, полупродуктов или катализатора, поэтому точная форма кинетического уравнения определяется экспериментально, или на основании анализа предполагаемого механизма реакции. Как правило, скорость сложной химической реакции определяется скоростью его самого медленного этапа (лимитирующей стадии).
3. Давление
Концентрация газов напрямую зависит от давления . При повышении давления повышается концентрация газов. Математическое выражение этой зависимости (для идеального газа) — уравнение Менделеева-Клапейрона:
pV = νRT
Таким образом, если среди реагентов есть газообразное вещество, то при повышении давления скорость химической реакции увеличивается, при понижении давления — уменьшается .
Например. Как изменится скорость реакции сплавления извести с оксидом кремния:
при повышении давления?
Правильным ответом будет – никак, т.к. среди реагентов нет газов, а карбонат кальция – твердая соль, нерастворимая в воде, оксид кремния – твердое вещество. Газом будет продукт – углекислый газ. Но продукты не влияют на скорость прямой реакции.
4. Катализатор
Еще один способ увеличить скорость химической реакции – направить ее по другому пути, заменив прямое взаимодействие, например, веществ А и В серией последовательных реакций с третьим веществом К, которые требуют гораздо меньших затрат энергии (имеют более низкий активационный энергетический барьер) и протекают при данных условиях быстрее, чем прямая реакция. Это третье вещество называют катализатором .

Катализаторы – это химические вещества, участвующие в химической реакции, изменяющие ее скорость и направление, но не расходующиеся в ходе реакции (по окончании реакции не изменяющиеся ни по количеству, ни по составу). Примерный механизм работы катализатора для реакции вида А + В можно представить так:
A + K = AK
AK + B = AB + K
Процесс изменения скорости реакции при взаимодействии с катализатором называют катализом. Катализаторы широко применяют в промышленности, когда необходимо увеличить скорость реакции, либо направить ее по определенному пути.
По фазовому состоянию катализатора различают гомогенный и гетерогенный катализ.
Гомогенный катализ – это когда реагирующие вещества и катализатор находятся в одной фазе (газ, раствор). Типичные гомогенные катализаторы – кислоты и основания. органические амины и др.
Гетерогенный катализ – это когда реагирующие вещества и катализатор находятся в разных фазах. Как правило, гетерогенные катализаторы – твердые вещества. Т.к. взаимодействие в таких катализаторах идет только на поверхности вещества, важным требованием для катализаторов является большая площадь поверхности. Гетерогенные катализаторы отличает высокая пористость, которая увеличивает площадь поверхности катализатора. Так, суммарная площадь поверхности некоторых катализаторов иногда достигает 500 квадратных метров на 1 г катализатора. Большая площадь и пористость обеспечивают эффективное взаимодействие с реагентами. К гетерогенным катализаторам относятся металлы, цеолиты — кристаллические минералы группы алюмосиликатов (соединений кремния и алюминия), и другие.

Пример гетерогенного катализа – синтез аммиака:
В качестве катализатора используется пористое железо с примесями Al2O3 и K2O.
Сам катализатор не расходуется в ходе химической реакции, но на поверхности катализатора накапливаются другие вещества, связывающие активные центры катализатора и блокирующие его работу (каталитические яды). Их необходимо регулярно удалять, путем регенерации катализатора.
В биохимических реакция очень эффективными оказываются катализаторы – ферменты. Ферментативные катализаторы действуют эффективно и избирательно, с избирательностью 100%. К сожалению, ферменты очень чувствительны к повышению температуры, кислотности среды и другим факторам, поэтому есть ряд ограничений для реализации в промышленных масштабах процессов с ферментативным катализом.
Катализаторы не стоит путать с инициаторами процесса и ингибиторами.
Например , для инициирования радикальной реакции хлорирования метана необходимо облучение ультрафиолетом. Это не катализатор. Некоторые радикальные реакции инициируются пероксидными радикалами. Это также не катализаторы.
Ингибиторы – это вещества, которые замедляют химическую реакцию. Ингибиторы могут расходоваться и участвовать в химической реакции. При этом ингибиторы не являются катализаторами наоборот. Обратный катализ в принципе невозможен – реакция в любом случае будет пытаться идти по наиболее быстрому пути.
5. Площадь соприкосновения реагирующих веществ
Для гетерогенных реакций одним из способов увеличить число эффективных соударений является увеличение площади реакционной поверхности . Чем больше площадь поверхности контакта реагирующих фаз, тем больше скорость гетерогенной химической реакции. Порошковый цинк гораздо быстрее растворяется в кислоте, чем гранулированный цинк такой же массы.
В промышленности для увеличения площади контактирующей поверхности реагирующих веществ используют метод «кипящего слоя».
Например , при производстве серной кислоты методом «кипящего слоя» производят обжиг колчедана.
6. Природа реагирующих веществ
На скорость химических реакций при прочих равных условиях также оказывают влияние химические свойства, т.е. природа реагирующих веществ.
Менее активные вещества будут имеют более высокий активационный барьер, и вступают в реакции медленнее, чем более активные вещества.
Более активные вещества имеют более низкую энергию активации, и значительно легче и чаще вступают в химические реакции.
Более стабильные вещества — это, например, те вещества, которые окружают нас в быту, либо существуют в природе.
Например , хлорид натрия NaCl (поваренная соль), или воды H2O, или металлическое железо Fe.
Более активные вещества мы можем встретить в быту и природе сравнительно редко.
Например , оксид натрия Na2O или сам натрий Na в быту и в природе не не встречаем, т.к. они активно реагируют с водой.
При небольших значениях энергии активации (менее 40 кДж/моль) реакция проходит очень быстро и легко. Значительная часть столкновений между частицами заканчивается химическим превращением. Например, реакции ионного обмена происходят при обычных условиях очень быстро.
При высоких значениях энергии активации (более 120 кДж/моль) лишь незначительное число столкновений заканчивается химическим превращением. Скорость таких реакций пренебрежимо мала. Например, азот с кислородом практически не взаимодействует при нормальных условиях.
При средних значениях энергии активации (от 40 до 120 кДж/моль) скорость реакции будет средней. Такие реакции также идут при обычных условиях, но не очень быстро, так, что их можно наблюдать невооруженным глазом. К таким реакциям относятся взаимодействие натрия с водой, взаимодействие железа с соляной кислотой и др.
Вещества, стабильные при нормальных условиях, как правило, имеют высокие значения энергии активации.
ХИМИЧЕСКАЯ КИНЕТИКА
ХИМИЧЕСКАЯ КИНЕТИКА – (от греч. кинетикос – движущий) наука о механизмах химических реакций и закономерностях их протекания во времени.
В 19 в. результате развития основ химической термодинамики химики научились рассчитывать состав равновесной смеси для обратимых химических реакций. Кроме того, на основании несложных расчетов можно было, не проводя экспериментов, сделать вывод о принципиальной возможности или невозможности протекания конкретной реакции в данных условиях. Однако «принципиальная возможность» реакции еще не означает, что она пойдет. Например, реакция С + О2 → СО2 с точки зрения термодинамики весьма благоприятна, во всяком случае, при температурах ниже 1000° С (при более высоких температурах происходит уже распад молекул СО2), т.е. углерод и кислород должны (практически со 100%-ным выходом) превратиться в диоксид углерода. Однако опыт показывает, что кусок угля может годами лежать на воздухе, при свободном доступе кислорода, не претерпевая никаких изменений. То же можно сказать и о множестве других известных реакций. Например, смеси водорода с хлором или с кислородом могут сохраняться очень долго без всяких признаков химических реакций, хотя в обоих случаях реакции термодинамически благоприятны. Это означает, что после достижения равновесия в стехиометрической смеси H2 + Cl2 должен остаться только хлороводород, а в смеси 2Н2 + О2 – только вода. Другой пример: газообразный ацетилен вполне стабилен, хотя реакция C2H2 → 2C + H2 не только термодинамически разрешена, но и сопровождается значительным выделением энергии. Действительно, при высоких давлениях, ацетилен взрывается, однако в обычных условиях он вполне стабилен.
Термодинамически разрешенные реакции, подобные рассмотренным, могут пойти только в определенных условиях. Например, после поджигания уголь или сера самопроизвольно соединяются с кислородом; водород легко реагирует с хлором при повышении температуры или при действии ультрафиолетового света; смесь водорода с кислородом (гремучий газ) взрывается при поджигании или при внесении катализатора. Почему же для осуществления всех этих реакций необходимы специальные воздействия – нагревание, облучение, действие катализаторов? Химическая термодинамика не дает ответа на этот вопрос – понятие времени в ней отсутствует. В то же время для практических целей очень важно знать, пройдет ли данная реакция за секунду, за год или же за многие тысячелетия.

Опыт показывает, что скорость разных реакций может отличаться очень сильно. Практически мгновенно идут многие реакции в водных растворах. Так, при добавлении избытка кислоты к щелочному раствору фенолфталеина малинового цвета раствор мгновенно обесцвечивается, это означает, что реакция нейтрализации, а также реакция превращения окрашенной формы индикатора в бесцветную идут очень быстро. Значительно медленнее идет реакция окисления водного раствора иодида калия кислородом воздуха: желтая окраска продукта реакции – иода появляется лишь через продолжительное время. Медленно протекают процессы коррозии железных и особенно медных сплавов, многие другие процессы.
Предсказание скорости химической реакции, а также выяснение зависимости этой скорости от условий проведения реакции – одна из важных задач химической кинетики – науки, изучающей закономерности протекания реакций во времени. Не менее важна и вторая задача, стоящая перед химической кинетикой – изучение механизма химических реакций, то есть детального пути превращения исходных веществ в продукты реакции.
Скорость реакции.
Проще всего определить скорость для реакции, протекающей между газообразными или жидкими реагентами в гомогенной (однородной) смеси в сосуде постоянного объема. В этом случае скорость реакции определяется как изменение концентрации любого из участвующих в реакции веществ (это может быть исходное вещество или продукт реакции) в единицу времени. Это определение можно записать в виде производной: v = dc/dt, где v – скорость реакции; t – временя, c – концентрация. Эту скорость легко определить, если есть экспериментальные данные по зависимости концентрации вещества от времени. По этим данным можно построить график, который называется кинетической кривой. Скорость реакции в заданной точке кинетической кривой определяется наклоном касательной в этой точке. Определение наклона касательной всегда связано с некоторой ошибкой. Точнее всего определяется начальная скорость реакции, поскольку вначале кинетическая кривая обычно близка к прямой; это облегчает проведение касательной в начальной точке кривой.
Если время измерять в секундах, а концентрацию – в молях на литр, то скорость реакции измеряется в единицах моль/(л·с). Таким образом, скорость реакции не зависит от объема реакционной смеси: при одинаковых условиях она будет одинаковой и в маленькой пробирке, и в многотоннажном реакторе.
Величина dt всегда положительна, тогда как знак dc зависит от того, как изменяется со временем концентрация – уменьшается (для исходных веществ) или увеличивается (для продуктов реакции). Чтобы скорость реакции всегда оставалась величиной положительной, в случае исходных веществ перед производной ставят знак минус: v = –dc/dt. Если реакция идет в газовой фазе, вместо концентрации веществ в уравнении скорости часто используют давление. Если газ близок к идеальному, то давление р связано с концентрацией с простым уравнением: p = cRT.
В ходе реакции разные вещества могут расходоваться и образовываться с разной скоростью, в соответствии с коэффициентами в стехиометрическом уравнении (см. СТЕХИОМЕТРИЯ), поэтому, определяя скорость конкретной реакции, следует учитывать эти коэффициенты. Например, в реакции синтеза аммиака 3H2 + N2 → 2NH3 водород расходуется в 3 раза быстрее, чем азот, а аммиак накапливается в 2 раза быстрее, чем расходуется азот. Пэтому уравнение скорости для этой реакции записывают следующим образом: v = –1/3 dp(H2)/dt = –dp(N2)/dt = +1/2 dp(NH3)/dt. В общем случае, если реакция стехиометрическая, т.е. протекает точно в соответствии с записанным уравнением: aA + bB → cC + dD, ее скорость определяют как v = –(1/a)d[A]/dt = –(1/b)d[B]/dt = (1/c)d[C]/dt = (1/d)d[D]/dt (квадратными скобками принято указывать молярную концентрацию веществ). Таким образом, скорости по каждому веществу жестко связаны между собой и, определив экспериментально скорость для любого участника реакции, легко рассчитать ее для любого другого вещества.
Большинство реакций, используемых в промышленности, относятся к гетерогенно-каталитическим. Они протекают на поверхности раздела фаз между твердым катализатором и газовой или жидкой фазой. На поверхности раздела двух фаз протекают и такие реакции как обжиг сульфидов, растворение металлов, оксидов и карбонатов в кислотах, ряд других процессов. Для таких реакций скорость зависит и от величины поверхности раздела, поэтому скорость гетерогенной реакции относят не к единице объема, а к единице поверхности. Измерить величину поверхности, на которой идет реакция, не всегда просто.
Если реакция протекает в замкнутом объеме, то ее скорость в большинстве случаев максимальна в начальный момент времени (когда максимальна концентрация исходных веществ), а затем, по мере превращения исходных реагентов в продукты и, соответственно, снижения их концентрации, скорость реакции уменьшается. Встречаются и реакции, в которых скорость увеличивается со временем. Например, если медную пластинку опустить в раствор чистой азотной кислоты, то скорость реакции будет расти со временем, что легко наблюдать визуально. Ускоряются со временем также процессы растворения алюминия в растворах щелочей, окисления многих органических соединений кислородом, ряд других процессов. Причины такого ускорения могут быть разными. Например, это может быть связано с удалением защитной оксидной пленки с поверхности металла, или с постепенным разогревом реакционной смеси, или с накоплением веществ, ускоряющих реакцию (такие реакции называются автокаталитическими).
В промышленности реакции обычно проводят путем непрерывной подачи в реактор исходных веществ и вывода продуктов. В таких условиях можно добиться постоянной скорости протекания химической реакции. С постоянной скоростью протекают и фотохимические реакции при условии полного поглощения падающего света (см. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ).
Лимитирующая стадия реакции.
Если реакция осуществляется путем последовательно протекающих стадий (не обязательно все из них являются химическими) и одна из этих стадий требует значительно большего времени, чем остальные, то есть идет намного медленнее, то такая стадия называется лимитирующей. Именно эта самая медленная стадия определяет скорость всего процесса. Рассмотрим в качестве примера каталитическую реакцию окисления аммиака. Здесь возможны два предельных случая.
1. Поступление молекул реагентов – аммиака и кислорода к поверхности катализатора (физический процесс) происходит значительно медленнее, чем сама каталитическая реакция на поверхности. Тогда для повышения скорости образования целевого продукта – оксида азота совершенно бесполезно повышать эффективность катализатора, а надо позаботиться об ускорении доступа реагентов к поверхности.
2. Подача реагентов к поверхности происходит значительно быстрее самой химической реакции. Вот здесь имеет смысл совершенствовать катализатор, подбирать оптимальные условия для каталитической реакции, так как лимитирующей стадией в данном случае является каталитическая реакция на поверхности.
Теория столкновений.
Исторически первой теорией, на основании которой можно было рассчитывать скорости химических реакций, была теория столкновений. Очевидно, что для того, чтобы молекулы прореагировали, они прежде всего должны столкнуться. Отсюда следует, что реакция должна идти тем быстрее, чем чаще сталкиваются друг с другом молекулы исходных веществ. Поэтому каждый фактор, влияющий на частоту столкновений между молекулами, будет влиять и на скорость реакции. Некоторые важные закономерности, касающиеся столкновений между молекулами, были получены на основании молекулярно-кинетической теории газов.
В газовой фазе молекулы движутся с большими скоростями (сотни метров в секунду) и очень часто сталкиваются друг с другом. Частота столкновений определяется прежде всего числом частиц в единице объема, то есть концентрацией (давлением). Частота столкновений зависит также и от температуры (с ее повышением молекулы движутся быстрее) и от размера молекул (большие молекулы сталкиваются друг с другом чаще, чем маленькие). Однако концентрация влияет на частоту столкновений значительно сильнее. При комнатной температуре и атмосферном давлении каждая молекула средних размеров испытывает в секунду несколько миллиардов столкновений.
На основании этих данных можно рассчитать скорость реакции А + В → С между двумя газообразными соединениями А и В, предполагая, что химическая реакция проходит при каждом столкновении молекул реагентов. Пусть в литровой колбе при атмосферном давлении есть смесь реагентов А и В при равных концентрациях. Всего в колбе будет 6·10 23 /22,4 = 2,7·10 22 молекул, из которых 1,35·10 22 молекул вещества А и столько же молекул вещества В. Пусть за 1 с каждая молекула А испытывает 10 9 столкновений с другими молекулами, из которых половина (5·10 8 ) приходится на столкновения с молекулами В (столкновения А + А не приводят к реакции). Тогда всего в колбе за 1 с происходит 1,35·10 22 ·5·10 8
7·10 30 столкновений молекул А и В. Очевидно, что если бы каждое из них приводило к реакции, она прошла бы мгновенно. Однако многие реакции идут достаточно медленно. Отсюда можно сделать вывод, что лишь ничтожная доля столкновений между молекулами реагентов приводит к взаимодействию между ними.
Для создания теории, которая позволяла бы рассчитать скорость реакции на основании молекулярно-кинетической теории газов, нужно было уметь рассчитывать общее число столкновений молекул и долю «активных» столкновений, приводящих реакции. Нужно было также объяснить, почему скорость большинства химических реакций сильно возрастает при повышении температуры – скорость молекул и частота столкновений между ними увеличиваются с температурой незначительно – пропорционально 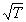 , то есть всего в 1,3 раза при повышении температуры от 293 К (20° С) до 373 К (100° С), тогда как скорость реакции при этом может увеличиться в тысячи раз.
, то есть всего в 1,3 раза при повышении температуры от 293 К (20° С) до 373 К (100° С), тогда как скорость реакции при этом может увеличиться в тысячи раз.
Эти проблемы были решены на основании теории столкновений следующим образом. При столкновениях молекулы непрерывно обмениваются скоростями и энергиями. Так, данная молекула в результате «удачного» столкновения может заметно увеличить свою скорость, тогда как при «неудачном» столкновении она может почти остановиться (похожую ситуацию можно наблюдать на примере бильярдных шаров). При нормальном атмосферном давлении столкновения, а следовательно, изменения скорости происходят с каждой молекулой миллиарды раз в секунду. При этом скорости и энергии молекул в значительной степени усредняются. Если в данный момент времени «пересчитать» в заданном объеме газа молекулы, обладающие определенными скоростями, то окажется, что значительная часть их имеет скорость, близкую к средней. В то же время многие молекулы обладают скоростью меньше средней, а часть движется со скоростями больше средней. С увеличением скорости доля молекул, имеющих данную скорость, быстро уменьшается. В соответствии с теорией столкновений, реагируют только те молекулы, которые при столкновении обладают достаточно высокой скоростью (и, следовательно, большим запасом кинетической энергии). Такое предположение было сделано в 1889 году шведским химиком Сванте Аррениусом.
Энергия активации.
Аррениус ввел в обиход химиков очень важное понятие энергии активации (Ea) – это та минимальная энергия, которой должна обладать молекула (или пара реагирующих молекул), чтобы вступить в химическую реакцию. Энергию активации измеряют обычно в джоулях и относят не к одной молекуле (это очень маленькая величина), а к молю вещества и выражают в единицах Дж/моль или кДж/моль. Если энергия сталкивающихся молекул меньше энергии активации, то реакция не пойдет, а если равна или больше, то молекулы прореагируют.
Энергии активации для разных реакций определяют экспериментально (из зависимости скорости реакции от температуры). Изменяться энергия активации может в довольно широких пределах – от единиц до нескольких сотен кДж/моль. Например, для реакции 2NO2 → N2O4 энергия активации близка к нулю, для реакции 2Н2О2 → 2Н2О + О2 в водных растворах Ea = 73 кДж/моль, для термического разложения этана на этилен и водород Ea = 306 кДж/моль.
Энергия активации большинства химических реакций значительно превышает среднюю кинетическую энергию молекул, которая при комнатной температуре составляет всего лишь около 4 кДж/моль и даже при температуре 1000° С не превышает 16 кДж/моль. Таким образом, чтобы прореагировать, молекулы обычно должны иметь скорость значительно больше средней. Например, в случае Ea = 200 кДж/моль сталкивающиеся молекулы небольшой молекулярной массы должны иметь скорость порядка 2,5 км/с (энергия активации в 25 раз больше средней энергии молекул при 20° С). И это – общее правило: для большинства химических реакций энергия активации значительно превышает среднюю кинетическую энергию молекул.
Вероятность для молекулы запасти в результате серии столкновений большую энергию очень мала: такой процесс требует для нее колоссального числа последовательных «удачных» столкновений, в результате которых молекула только набирает энергию, не теряя ее. Поэтому для многих реакций лишь ничтожная доля молекул имеет энергию, достаточную для преодоления барьера. Эта доля, в соответствии с теорией Аррениуса, определяется формулой: a = e –Ea /RT = 10 –Ea /2,3RT
10 –Ea /19Т, где R = 8,31 Дж/(моль . К). Из формулы следует, что доля молекул, обладающих энергией Ea, как и доля активных столкновений a, очень сильно зависит как от энергии активации, так и от температуры. Например, для реакции с Ea = 200 кДж/моль при комнатной температуре (Т
300 К) доля активных столкновений ничтожно мала: a = 10 –200000/(19,300)
10 –35 . И если каждую секунду в сосуде происходит 7·10 30 столкновений молекул А и В, то понятно, что реакция идти не будет.
Если увеличить вдвое абсолютную температуру, т.е. нагреть смесь до 600 К (327° С); при этом доля активных столкновений резко возрастет: a = 10 –200000/(19,600)
4·10 –18 . Таким образом, повышение температуры в 2 раза увеличило долю активных столкновений в 4·10 17 раз. Теперь каждую секунду из общего числа примерно 7·10 30 столкновений к реакции будет приводить 7·10 30 ·4·10 –18
3·10 13 . Такая реакция, в которой каждую секунду исчезает 3·10 13 молекул (из примерно 10 22 ), хотя и очень медленно, но все же идет. Наконец, при температуре 1000 К (727° C) a
3·10 –11 (из каждых 30 миллиардов столкновений данной молекулы реагента одно приводит к реакции). Это уже много, так как за 1 с в реакцию будут вступать 7·10 30 ·3·10 –11 = 2·10 20 молекул, и такая реакция пройдет за несколько минут (с учетом снижения частоты столкновений с уменьшением концентрации реагентов).
Теперь понятно, почему повышение температуры может так сильно увеличить скорость реакции. Средняя скорость (и энергия) молекул с повышением температуры увеличивается незначительно, но зато резко повышается доля «быстрых» (или «активных») молекул, обладающих достаточной для протекания реакции скоростью движения или достаточной колебательной энергией.
Расчет скорости реакции с учетом общего числа столкновений и доли активных молекул (т.е. энергии активации), часто дает удовлетворительное соответствие с экспериментальными данными. Однако для многих реакций наблюдаемая на опыте скорость оказывается меньше рассчитанной по теории столкновений. Это объясняется тем, что для осуществления реакции нужно, чтобы столкновение было удачным не только энергетически, но и «геометрически», то есть молекулы должны в момент столкновения определенным образом ориентироваться относительно друг друга. Таким образом, при расчетах скорости реакций по теории столкновений, помимо энергетического, учитывают и стерический (пространственный) фактор для данной реакции.
Уравнение Аррениуса.
Зависимость скорости реакции от температуры обычно описывают уравнением Аррениуса, которое в простейшем виде можно записать как v = v0 a = v0e –Ea/RT , где v0 – скорость, которую имела бы реакция при нулевой энергии активации (фактически это частота столкновений в единице объеме). Поскольку v0 слабо зависит от температуры, все определяет второй сомножитель – экспоненциальный: с увеличением температуры этот сомножитель быстро увеличивается, причем тем быстрее, чем больше энергия активации Еа. Указанная зависимость скорости реакции от температуры называется уравнением Аррениуса, оно – одно из важнейших в химической кинетике. Для приблизительной оценки влияния температуры на скорость реакции иногда используют так называемое «правило Вант-Гоффа» (см. ВАНТ-ГОФФА ПРАВИЛО).
Если реакция подчиняется уравнению Аррениуса, логарифм ее скорости (измеренной, например, в начальный момент) должен линейно зависеть от абсолютной температуры, то есть график зависимости lnv от 1/Т должен быть прямолинейным. Наклон этой прямой равен энергии активации реакции. По такому графику можно предсказать, какова будет скорость реакции при данной температуре или же – при какой температуре реакция будет идти с заданной скоростью.
Несколько практических примеров использования уравнения Аррениуса.
1. На упаковке замороженного продукта написано, что его можно хранить на полке холодильника (5° С) в течение суток, в морозильнике, отмеченном одной звездочкой (–6° С), – неделю, двумя звездочками (–12° С) – месяц, а в морозильнике со значком *** (что означает температуру в нем –18° С) – 3 месяца. Предположив, что скорость порчи продукта обратно пропорциональна гарантийному сроку хранения tхр, в координатах lntхр, 1/Т получаем, в соответствии с уравнением Аррениуса, прямую. Из нее можно рассчитать энергию активации биохимических реакций, приводящие к порче данного продукта (около 115 кДж/моль). Из того же графика можно выяснить, до какой температуры надо охладить продукт, чтобы его можно было хранить, например, 3 года; получается –29° С.
2. Альпинисты знают, что в горах трудно сварить яйцо, и вообще любую пищу, требующую более или менее длительного кипячения. Качественно причина этого понятна: с понижением атмосферного давления уменьшается температура кипения воды. С помощью уравнения Аррениуса можно рассчитать, сколько времени потребуется, например, чтобы сварить вкрутую яйцо в г. Мехико, расположенном на высоте 2265 м, где нормальным считается давление 580 мм рт.ст., а вода при таком пониженном давлении кипит при 93° С. Энергия активации реакция «свертывания» (денатурации) белка была измерена и оказалась очень большой по сравнению со многими другими химическими реакциями – порядка 400 кДж/моль (она может несколько отличаться для различных белков). В таком случае понижение температуры от 100 до 93° С (то есть от 373 до 366 К) приведет к замедлению реакции в 10 (400000/19)(1/366 – 1/373) = 11,8 раза. Именно поэтому жители высокогорья предпочитают варке пищи ее жарку: температура сковородки, в отличие от температуры кастрюли с кипятком, не зависит от атмосферного давления.
3. В кастрюле-скороварке пища готовится при повышенном давлении и, следовательно, при повышенной температуре кипения воды. Известно, что в обычной кастрюле говядина варится 2–3 часа, а компот из яблок – 10–15 мин. Учитывая, что оба процесса имеют близкую энергию активации (около 120 кДж/моль), можно по уравнению Аррениуса рассчитать, что в скороварке при 118°С мясо будет вариться 25–30 мин, а компот – всего 2 мин.
Уравнение Аррениуса очень важно для химической промышленности. При протекании экзотермической реакции выделяющаяся тепловая энергия нагревает не только окружающую среду, но и сами реагенты. это может привести к нежелательному сильному ускорению реакции. Расчет изменения скорости реакции и скорости тепловыделения при повышении температуры позволяет избежать теплового взрыва (см. ВЗРЫВЧАТЫЕ ВЕЩЕСТВА).
Зависимость скорости реакции от концентрации реагентов.
Скорость большинства реакций со временем постепенно снижается. Этот результат хорошо согласуется с теорией столкновений: по мере протекания реакции концентрации исходных веществ падают, снижается и частота столкновений между ними; соответственно уменьшается и частота столкновения активных молекул. Это приводит к уменьшению скорости реакции. В этом состоит сущность одного из основных законов химической кинетики: скорость химической реакции пропорциональна концентрации реагирующих молекул. Математически это можно записать в виде формулы v = k[A][B], где k – постоянная, называемая константой скорости реакции. Приведенное уравнение называется уравнением скорости химической реакции или кинетическим уравнением. Константа скорости для данной реакции не зависит от концентрации реагентов и от времени, но она зависит от температуры в соответствии с уравнением Аррениуса: k = k0e –Ea/RT .
Простейшее уравнение скорости v = k[A][B] всегда справедливо в том случае, когда молекулы (или другие частицы, например, ионы) А, сталкиваясь с молекулами В, могут непосредственно превращаться в продукты реакции. Подобные реакции, идущие в один прием (как говорят химики, в одну стадию), называются элементарными реакциями. Таких реакций немного. Большинство реакций (даже таких с виду таких простых как H2 + I2 ® 2HI) не являются элементарными, поэтому исходя из стехиометрического уравнения такой реакции записать его кинетическое уравнение нельзя.
Кинетическое уравнение можно получить двумя способами: экспериментально – измеряя зависимость скорости реакции от концентрации каждого реагента по отдельности, и теоретически – если известен детальный механизм реакции. Чаще всего (но не всегда) кинетическое уравнение имеет вид v = k[A] x [B] y , где x и y называются порядками реакции по реагентам А и В. Эти порядки, в общем случае, могут быть целыми и дробными, положительными и даже отрицательными. Например, кинетическое уравнение для реакции термического распада ацетальдегида CH3CHO ® CH4 + CO имеет вид v = k[CH3CHO] 1,5 , т.е. реакция имеет полуторный порядок. Иногда возможно случайное совпадение стехиометрических коэффициентов и порядков реакции. Так, эксперимент показывает, что реакция H2 + I2 ® 2HI имеет первый порядок как по водороду, так и по иоду, то есть ее кинетическое уравнение имеет вид v = k[H2][I2] (именно поэтому эту реакцию в течение многих десятилетий считали элементарной, пока в 1967 не был доказан ее более сложный механизм).
Если известно кинетическое уравнение, т.е. известно, как скорость реакции зависит от концентраций реагентов в каждый момент времени, и известна константа скорости, то можно рассчитать зависимость от времени концентраций реагентов и продуктов реакции, т.е. теоретически получить все кинетические кривые. Для таких расчетов используются методы высшей математики или компьютерные расчеты, и они не представляют принципиальных трудностей.
С другой стороны, полученное экспериментально кинетическое уравнение помогает судить о механизме реакции, т.е. о совокупности простых (элементарных) реакций. Выяснение механизмов реакций является важнейшей задачей химической кинетики. Это очень трудная задача, так как механизм даже простой с виду реакции может включать множество элементарных стадий.
Можно проиллюстрировать применение кинетических методов для определения механизма реакции на примере щелочного гидролиза алкилгалогенидов с образованием спиртов: RX + OH – → ROH + X – . Экспериментально было обнаружено, что для R = CH3, C2H5 и т.д. и X = Cl скорость реакции прямо пропорционально концентрациям реагентов, т.е. имеет первый порядок по галогениду RX и первый – по щелочи, и кинетическое уравнение имеет вид v = k1[RX][OH – ]. В случае третичных алкилиодидов (R = (CH3)3C, X = I) порядок по RX – первый, а по щелочи – нулевой: v = k2[RX]. В промежуточных случаях, например, для изопропилбромида (R = (CH3)2CH, X = Br), реакция описывается более сложным кинетическим уравнением: v = k1[RX][OH – ] + k2[RX]. На основании этих кинетических данных был сделан следующий вывод о механизмах подобных реакций.
В первом случае реакция идет в один прием, путем непосредственного столкновения молекул спирта с ионами ОН – (так называемый механизм SN2). Во втором случае реакция идет в две стадии. Первая стадия – медленная диссоциация алкилиодида на два иона: RI → R + + I – . Вторая – очень быстрая реакция между ионами: R + + OH – → ROH. Скорость суммарной реакции зависит только от медленной (лимитирующей) стадии, поэтому она не зависит от концентрации щелочи; отсюда – нулевой порядок по щелочи (механизм SN1). В случае вторичных алкилбромидов осуществляются одновременно оба механизма, поэтому кинетическое уравнение более сложное.
http://chemege.ru/kinetika/
http://www.krugosvet.ru/enc/nauka_i_tehnika/himiya/HIMICHESKAYA_KINETIKA.html