Уравнение аррениуса в интегральной форме и дифференциальной форме
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Конспект лекций для студентов биофака ЮФУ (РГУ)
2.1 СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
2.1.9 Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Я. Г. Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ . Математически правило Вант-Гоффа можно записать следующим образом:
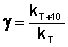 (II.29)
(II.29)
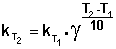 (II.30)
(II.30)
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
2.1.10 Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. С. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом:
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е’А выше, нежели энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
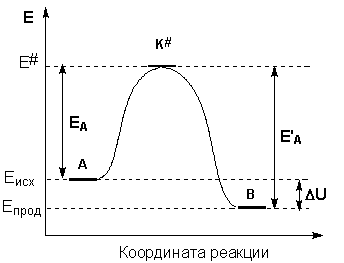
Рис. 2.5 Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.6):
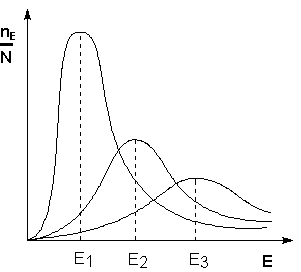
Рис. 2.6 Распределение частиц по энергии
Здесь nЕ/N – доля частиц, обладающих энергией E;
Ei — средняя энергия частиц при температуре Ti (T1 уравнения Аррениуса . Согласно уравнению изобары Вант-Гоффа,
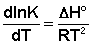 (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
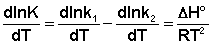 (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
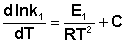 (II.33)
(II.33)
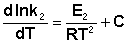 (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации :
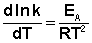 (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
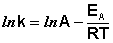 (II.36)
(II.36)
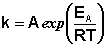 (II.37)
(II.37)
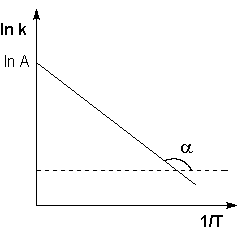
Рис. 2.7 Зависимость логарифма константы скорости химической
реакции от обратной температуры.
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
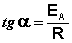 (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
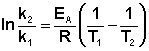 (II.39)
(II.39)
Copyright © С. И. Левченков, 1996 — 2005.
Уравнение Аррениуса в дифференциальной и интегральной формах. Энергия активации и предэкспоненциальный множитель, методы их нахождения из эксперимента.
Вопросы к экзамену по физической химии (2-я часть)
1. Теория диссоциации Аррениуса. Причины диссоциации. Сильные и слабые электролиты, степень и константа диссоциации, их зависимость от температуры, концентрации и типа растворителя. Закон разведения Оствальда. Ассоциация и сольватация ионов.
2. Электропроводность растворов сильных электролитов. Уравнение Кольрауша (закон квадратного корня). Электрофоретический и релаксационный эффекты. Влияние высокой напряженности поля и высокой частоты на электропроводность растворов.
3. Скорость движения и подвижность ионов. Закон Кольрауша (независимого движения ионов). Молярные электропроводности ионов при бесконечном разведении. Числа переноса. Эстафетный механизм переноса электричества ионами Н и ОН.
4.Проводники I и II рода, ионная и электронная проводимость. Удельная электропроводность и ее зависимость от разных факторов. Молярная (эквивал.) электропроводность. Зависимость проводимости от концентрации для сильных и для слабых электролитов.
5.Активности электролита и ионов, коэффициенты активности, их экспериментальное и теоретическое нахождение. Ионная сила раствора (определение). Правило ионной силы.
6. Определение степени и константы диссоциации слабых электролитов по измерениям электропроводности. Нахождение по этим данным ПР и термодинамических характеристик процесса диссоциации. Кондуктометрическое титрование.
7. Основные положения теории Дебая-Хюккеля. Предельный закон, 1-е и 2-е приближения. Зависимость коэф. активности от ионной силы.
8. Применение измерений ЭДС для нахождения термодинамических характеристик реакций в цепи, рН, ПР, констант равновесия. Примеры.
9. Типы гальванических элементов: химические концентрационные, с переносом, без переноса. Запись полуреакций.Диффузионный потенциал и его устранение.
10.Электроды 1 и 2 рода, газовые, окислительно-восстановительные. Уравнение Нернста для разных типов электродов. Водородный электрод.
11. Зависимость ЭДС гальванического элемента от температуры, уравнение Гиббса –Гельмгольца. Нахождение энтропии реакции по этим данным.
12. Условный электродный потенциал (потенциал в водородной шкале). Связь ЭДС цепи с потенциалами электродов. Запись цепей, правила знаков ЭДС и электродных потенциалов.
13. Электрохимические цепи. Возникновение скачка потенциалов на границе проводников 1 и 2 рода. Гальвани-потенциал. Двойной слой и его строение.
14. Термодинамика электрохимических цепей. Вывод и анализ уравнения Нернста в общем виде и в частных случаях.
15. Скорость хим. реакции, методы определения скорости, молекулярность, порядок (частный и общий) время полупревращения, константа скорости и ее размерность. Зависимость скорости р-ций от температуры. Правило Вант-Гоффа и его обоснование.
16. Частный и общий порядки реакции. Дифференциальные и интегральные методы определения порядка реакции. Концентрационный и временной порядки.
17. Теория переходного состояния (ТПС), ее постулаты, квазитермодинамическая форма уравнения ТПС, энтальпия и энтропия активации, трансмиссионный коэффициент, связь энтальпии акт. с экспериментальной (эффективной) энергией активации.
18. Теория переходного состояния (ТПС), поверхность потенциальной энергии, путь реакции, координата реакции, энергетический барьер и переходное состояние, истинная энергия активации элементарной реакции.
19. Сложные реакции, принцип независимости протекания элементарных реакций. Параллельные р-ции 1-го порядка, их дифференциальные и интегральные кинетические уравнения. Обратимые р-ции 1-го порядка, их уравнения.
20. Методы приближенного установления кинетики сложных р-ций: метод стационарных концентраций, принцип лимитирующей стадии, принцип микрообратимости.
21. Механизм мономолекулярных газовых реакций, схема Линдемана. Причины изменения порядка с 1-го на 2-й при изменении давления.
Уравнение Аррениуса в дифференциальной и интегральной формах. Энергия активации и предэкспоненциальный множитель, методы их нахождения из эксперимента.
23. Реакции нулевого, первого и второго порядков, их уравнения и кинетические кривые. Линеаризация кинетических кривых, время полупревращения.
24.Теория активных соударений (ТАС). Константа скорости бимолекулярной реакции. Физический смысл предэкспоненты и энергии активации в рамках ТАС. Стерический фактор.
25. Простые и сложные реакции. Последовательные реакции 1-го порядка. Система уравнений, описывающих кинетику. Кинетические кривые участников. Время достижения максимальной концентрации промежуточного вещества.
26. Реакции в растворах. Уравнение Бренстеда-Бьеррума. Кинетические особенности реакции Меншуткина.
27. Катализ, основные понятия. Каталитическая активность, удельная кат. активность. Гомогенный и гетерогенный катализ, автокатализ, влияние катализатора на термодинамические и кинетические характеристики р-ций.
28. Селективность катализаторов. Слитный и стадийный механизмы каталитических реакций. Энергетические диаграммы взаимодействия реагентов с катализатором.
29. Стационарный и нестационарный режимы протекания р-ций. Разветвленные реакции, предельные явления в них, пределы воспламенения (взрыва) цепной р-ции, роль т-ры и давления, полуостров и мыс воспламенения.
30.Цепные реакции, примеры. Звено цепи, длина цепи. Разветвленные и неразветвленные реакции, кинетика неразветвленных цепных реакций. Зарождение, развитие и обрыв цепи (линейный и квадратичный).
31. Фотохимические реакции. Механизм активации. Первичные и вторичные фотохим. процессы. Сенсибилизаторы и сенсибилизированные реакции. Квантовый выход.
32. Законы фотохимии. Квантовый выход. Кинетика процессов, происходящих с участием фотовозбужденных молекул. Различия реакций с фотохимическим и с термическим инициированием.
33. Диаграмма энергетических уровней молекул. Фотохимические и фотофизические процессы.
Уравнение Аррениуса в интегральной форме
где k-константа скорости реакции
e —основание скорости логарифма
R-универсальная газовая постоянная
T— температура по шкале Кельвина
2) Возрастание скорости реакции с ростом температуры принято характеризовать температурным коэффициентом скорости реакции , числом, показывающим, во сколько раз возрастает скорость данной реакции при повышении температуры системы на 10°С. Температурный коэффициент различных реакций различен.
3)При обычных температурах его значение для большинства реакций находится в пределах от 2. 4.
№2 Математическое выражение правила Вант Гоффа:
При повышении температуры на каждые 10 градусов константа скорости гомогенной элементарной реакции увеличивается в два — четыре раза.
 , где
, где  — скорость реакции при температуре
— скорость реакции при температуре  ,
,  — скорость реакции при температуре
— скорость реакции при температуре  ,
,  — температурный коэффициент реакции
— температурный коэффициент реакции
№3
Основа теории активных столкновений:
Для того чтобы произошла химическая реакция, молекулы реагентов должны:
1) столкнуться;
2) обладать достаточной энергией — энергией активации;
3) иметь благоприятную ориентацию для скорейшего взаимодействия друг с другом.
1. Энергияактивации реакции— это минимальное количество энергии, которое требуется сообщить системе, чтобы произошла реакция(выражается в джоулях на моль).

3. Переходный(активный) комплекс— конфигурация системы атомных ядер и электронов, участвующих в химической реакции, в момент преодоления системой энергетического барьера, разделяющего ее начальное и конечное состояния. (Теорию переходного состояния применяют для расчета скоростей химических реакций.)
4. Активные молекулы-это равновесие между нормальными молекулами и темпом. Только активные молекулы подвергаются химическому изменению. Если активные молекулы образуются эндотермически из нормальных молекул, то с повышением температуры должно происходить быстрое увеличение скорости химической реакции.
4)и5) 
2.2.3.Влияние_катализатора.
2) Типичными гомогенными катализаторами являются кислоты и основания. В качестве гетерогенных катализаторов применяются , металлы их оксиды и сульфиды. Реакции одного и того же типа могут протекать как с гомогенными, так и с гетерогенными катализаторами. Так, наряду с растворами кислот применяются имеющие кислотные свойства твёрдые Al2O3, TiO2, ThO2, алюмосиликаты, цеолиты. Гетерогенные катализаторы с основными свойствами: CaO, BaO, MgO.
Гетерогенные катализаторы имеют, как правило, сильно развитую поверхность, для чего их распределяют на инертном носителе (силикагель, оксид алюминия, активированный уголь и др.).
Катализатор обладает следующими свойствами :
1) Катализатор не влияет на общую стехиометрию реакции.
2) Катализатор одинаково ускоряет как прямую так и обратную реакции. Поэтому он увеличивает скорость достижения равновесия . Это означает, что он не меняет равновесные концентрации. Следовательно катализатор не изменяет выход реакции.
3) Катализатор влияет на механизм реакции, открывая новый путь протекания реакции. Энергия активации на этом новом пути оказывается ниже прежней, в результате оказывается, что большее число реагирующих молекул обладает энергией, необходимой для успешного столкновения. Соответственно этому увеличивается полное число столкновений, приводящих к реакции в единицу времени , а значит, увеличивается и скорость реакции.
4) Катализатор может увеличивать скорость одной реакции , но не увеличивать скорость другой, сходной реакции.
5) Катализатор принимает химическое участие в реакции. Он расходуется на одной стадии и регенерируется на следующей стадии реакции. Таким образом, катализатор может использоваться повторно, не подвергаясь окончательному превращению. Однако катализатор может изменять свое физическое состояние.
3)В химической промышленности катализаторы применяются весьма широко. Под влиянием катализаторов реакции могут ускоряться в миллионы раз и более. В некоторых случаях под действием катализаторов могут возбуждаться такие реакции, которые без них в данных условиях практически не протекают.
В отсутствии катализатора реакция протекает медленно.
2.2.4.
3)Химическое равновесие — состояние системы, в котором скорость прямой реакции равна скорости обратной реакции.
Константа равновесия—это есть безразмерная величина, которая являетсяколичественной характеристикой состояния химического равновесия, представляющей собой отношение равновесных концентраций продуктов реакции и исходных веществ, возведенных в степень их стехиометрических коэффициентов.
 , где K1- константа прямой и K2- константа обратной реакции
, где K1- константа прямой и K2- константа обратной реакции
Физический смысл константы равновесия заключается в том, что она показывает, во сколько раз скорость прямой реакции больше скорости обратной при данной температуре и концентрациях всех реагирующих веществ, равных 1 моль/л.
Константа равновесия зависит от природы реагирующих веществ и температуры. Изменения, происходящие в равновесной системе в результате внешних воздействий, определяются принципом подвижного равновесия (принцип Ле-Шателье): внешнее воздействие на систему, находящуюся в состоянии равновесия, приводит к смещению этого равновесия в направлении, при котором эффект произведенного воздействия ослабляется.

На смещение равновесия оказывает влияние:
1)изменение температуры: эндотермический процесс ускоряется в большей степени при повышении температуры и, наоборот, при понижении температуры ускоряется экзотермический процесс;
2)изменение давления(для реакций, протекающих в газовой фазе): при повышении давления равновесие реакции смещается в направлении образования веществ, занимающих меньший объем, и, наоборот, понижение давления способствует процессу, сопровождающемуся увеличением объема. Если реакция протекает без изменения объема, то изменение давления в системе не оказывает влияние на химическое равновесие.
3)изменение концентрации: увеличение концентрации исходных веществ приводит к увеличению скорости прямой реакции, при этом протекающий в системе процесс завершится, когда скорости прямой и обратной реакций станут равны и установится новое равновесие. Уменьшение концентрации одного из продуктов реакции (вывод из системы) приводит к смещению равновесия в сторону его образования.
4) катализатор не влияет константу химического равновесия, так как он снижает энергию активации прямой и обратной реакции на одну и ту же величину.
В заключение хочу сказать, что константа равновесия – это важная характеристика реакции. По её значению можно судить о направлении процесса при исходном соотношении концентраций реагирующих веществ, о максимально возможном выходе продукта при тех или иных условиях.
4)Закон действующих масс, которому подчиняется система в состоянии равновесия, гласит:
при постоянной температуре отношение произведения равновесных концентраций продуктов реакции, взятых в степенях, равных их коэффициентам, к произведению равновесных концентраций исходных веществ, взятых в степенях, равных их коэффициентам, есть величина постоянная.
В общем случае для обратимой реакции
aA + bB  dD + fF
dD + fF
в состоянии равновесия при постоянной температуре соблюдается соотношение

Постоянная величина (КС) называется константой равновесия данной реакции. Индекс » с» в обозначении этой величины показывает, что для расчета константы использовались концентрации.
Если константа равновесия велика, то равновесие сдвинуто в сторону продуктов прямой реакции, если мала, то — в сторону исходных веществ. Если константа равновесия очень велика, то говорят, что реакция » практически необратима», если константа равновесия очень мала, то реакция » практически не идет».
Константа равновесия — для каждой обратимой реакции величина постоянная только при постоянной температуре. Для одной и той же реакции при разных температурах константа равновесия принимает разные значения.
Приведенное выражение для закона действующих масс справедливо только для реакций, все участники которых представляют собой либо газы, либо растворенные вещества. В других случаях уравнение для константы равновесия несколько меняется. Приведенное выражение для закона действующих масс справедливо только для реакций, все участники которых представляют собой либо газы, либо растворенные вещества. В других случаях уравнение для константы равновесия несколько меняется.
Например, в протекающей при высокой температуре обратимой реакции
С (гр) + СО2  2СО (г)
2СО (г)
участвует твердый графит С (гр). Формально, пользуясь законом действующих масс, запишем выражение для константы равновесия этой реакции, обозначив ее К’:

Твердый графит, лежащий на дне реактора, реагирует только с поверхности, и его » концентрация» не зависит от массы графита и постоянна при любом соотношении веществ в газовой смеси.
Умножим правую и левую части уравнения на эту постоянную величину:
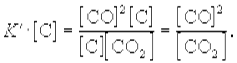
Получившаяся величина и есть константа равновесия этой реакции:

Аналогичным образом, для равновесия другой обратимой реакции, протекающей также при высокой температуре,
CaCO3(кр) СаО (кр) + СО2(г),
получим константу равновесия
В этом случае она просто равна равновесной концентрации углекислого газа.
То есть можно утверждать , что твердые исходные вещества и продукты реакции не влияют на смещение гетерогенного химического равновесия.
http://poisk-ru.ru/s2986t10.html
http://allrefrs.ru/2-4633.html