Практикум по электрохимии — часть 2

Концентрацию растворов сильных электролитов можно характеризовать
не только активностью а, но и ионной силой. Активность а показывает
концентрацию одного электролита в растворе, а через ионную силу можно
выражать концентрацию как одного, так и нескольких электролитов. Таким
образом, ионная сила показывает суммарную концентрацию всех
электролитов в растворе с учетом взаимодействия между ионами.
Ионная сила раствора (I, размерность концентрации) – это полусумма
произведений концентраций всех ионов в растворе на квадрат их заряда.
— моляльность i-го иона;
m – моляльность электролита; Z
заряд i-го иона.
Пример: раствор содержит 0,001 моль
РАСЧЕТ КОЭФФИЦИЕНТА АКТИВНОСТИ ПО ДЕБДШ И ХЮККЕЛЮ
В теории Дебая и Хюккеля расчет коэффициента активности данного сорта ионов базируется на следующих основных положениях.
Во-первых, принимается, что между ионами действуют только чисто электростатические (кулоновские) силы. Данное предположение оправдано для разбавленных растворов, поскольку кулоновские силы наиболее дальнодействующие и на больших расстояниях всеми другими видами взаимодействий между ионами можно пренебречь. В первоначальной теории ионы рассматривались как существующие в несольватированном виде. Это допущение не является справедливым, ибо в растворе каждый ион сольватирован и ион-дипольное взаимодействие между молекулами растворителя и ионом в не очень концентрированных растворах не зависит от концентрации. Поэтому вслед за Робинсоном и Стоксом правильно считать, что все дальнейшие расчеты относятся к ионам сольватированным.
Далее, в теории Дебая и Хюккеля принято, что число ионов в единице объема прямо пропорционально концентрации электролита, т. е. что при всех концентрациях электролит полностью диссоциирован. Если же диссоциация электролита неполная или ионы взаимодействуют с образованием нейтральных ионных пар, то в расчетных формулах должна фигурировать только концентрация электролита, распавшегося на ионы. При расчете кулоновского взаимодействия между ионами Дебай и Хюккель полагают, что в разбавленных растворах можно пользоваться значением диэлек- трической проницаемости чистого растворителя. Между тем, вблизи ионов молекулы растворителя ориентированы и, следовательно, диэлектрическая проницаемость такого структурированного слоя растворителя отличается от диэлектрической проницаемости того же растворителя с беспорядочным расположением молекул. Поэтому использование значения диэлектрической проницаемости чистого растворителя будет оправдано только для разбавленных растворов, когда вклад толщины структурированных слоев вокруг ионов в расстояние между ними незначителен.
В очень разбавленных растворах, когда центры ионов находятся друг от друга на столь больших расстояниях, что радиусом ионов можно пренебречь по сравнению с межионным расстоянием, ионы можно рассматривать как геометрические точки, несущие на себе электрический заряд.
Определение величины взаимодействия между ионами Дебай и Хюккель проводят на основе кристаллоподобной модели раствора электролита. Как в ионном кристалле вокруг любого произвольно выбранного положительно или отрицательно заряженного и имеется избыток ионов противоположного знака, так и в растворе вокруг любого выбранного (так называемого центрального) и имеется избыток ионов противоположного знака, образующих ионную атмосферу или ионное облако. В среднем заряд ионной атмосферы равен заряду центрального иона и по знаку противоположен ему. Однако ионное облако не является статическим образованием. Его нужно рассматривать как динамическое образование подвижных ионов, часть которых в каждую единицу времени покидает ионную атмосферу, заменяясь другими ионами. Но процесс этот происходит так, что суммарный электрический заряд ионного облака в среднем равен и противоположен знаку заряда центрального иона, т. е. в ионном облаке сохраняется избыток ионов, противоположных по знаку заряда центральному иону. Если центральный ион имеет валентность, скажем, +1, то суммарный избыточный заряд ионной атмосферы должен быть равен заряду электрона.
Такой заряд можно представить равномерно размытым по всему объему ионной атмосферы, если считать, что ионы ионной атмосферы участвуют в создании заряда ионного облака вокруг данного центрального иона только частью своих зарядов. Другая часть зарядов образует заряды ионных облаков вокруг других центральных ионов. Ведь на самом деле каждый ион, выбранный нами за центральный, в свою очередь включается в ионные облака других ионов, и все ионы в растворе в отношении своего центрального положения равноценны.
Расчет коэффициента активности по Дебаю и Хюккелю включает несколько этапов:
1. Определение плотности электрического заряда ионной атмосферы и ее связи с потенциалом ионной атмосферы.
2. Установление связи между потенциалом ионной атмосферы ее радиусом.
3. Расчет электрической работы образования ионной атмосферы
4. Установление связи между работой образования ионной
атмосферы и коэффициентом активности данного сорта ионов.
5. Вывод формулы для среднего ионного коэффициента активности.
На первом этапе расчета выберем начало координат, coвпадающее с положением центрального иона, и определим плотность электрического заряда в каком-нибудь бесконечно малом o6ъеме dv ионной атмосферы на расстоянии rот центрального иона (рис. 3.6). Введем обозначения:
ni — число ионов i-гo сорта в 1 м 3 раствора;
zi — заряд ионов i-го сорта с учетом их знака;
е — заряд электрона;
ψ — средний потенциал в объеме dv;
k — постоянная Больцмана; k = R/N, где R — газовая постоянная, а N — число молекул в моле (число Авогадро);
Рис. 3.6. Схема для расчета плотности заряда ионной атмосферы. Рис. Рис. 3.7. Положение точки Р в сферических координатах.
ε — диэлектрическая проницаемость растворителя;
ε0 — диэлектрическая проницаемость вакуума.
Для определения числа ионов i-гo сорта в объеме dv Дебай и Хюккель используют формулу Больцмана
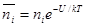
в которой 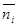 — число частиц в единице объема при воздействии на этот объем какого-либо непрерывного поля; ni — число частиц в той же единице объема в отсутствие поля; U — потенциальная энергия поля.
— число частиц в единице объема при воздействии на этот объем какого-либо непрерывного поля; ni — число частиц в той же единице объема в отсутствие поля; U — потенциальная энергия поля.
Для рассматриваемого случая формулу Больцмана записываем так:
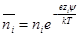
Электрический заряд i-го сорта ионов в объеме dv будет
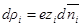
а в единице объема:
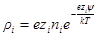
Для электролита или для смеси электролитов нужно произвести суммирование по всем сортам ионов с учетом знаков их заряда:
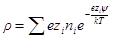
Чтобы в дальнейшем получить достаточно простое решение, приемлемое для разбавленных растворов, когда eziψ
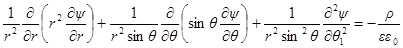
Поскольку ионная атмосфера обладает шаровой симметрией, то ∂ψ / ∂θ = 0и ∂ 2 ψ / ∂θ1 2 = 0, и
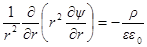
Подставим в уравнение Пуассона вместо ρ его выражение:
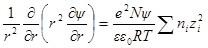
Умножим и разделим выражение в правой части на два и введем обозначение:
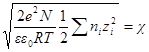
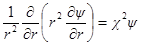
Для решения этого уравнения сперва продифференцируем по r выражение в скобках:
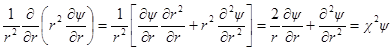
Введем теперь новую переменную ψr = Y и определим вторую производную Y по r:
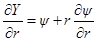
и 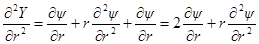
Сравнивая это выражение с предыдущим, получим:
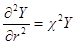
Из этого уравнения У определяется в виде
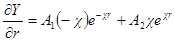
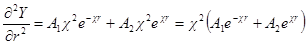
Так как Y= ψr, то
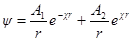
В этих уравнениях A1 и A2 — постоянные интегрирования. Константу A2 определяем из граничного условия, отвечающего r → ∞. Потенциал ψ при этом стремится к нулю, так как силы взаимодействия зарядов центрального иона и ионов в объеме dv с ростом r все более и более ослабевают. Это может быть лишь тогда, когда в уравнении для потенциала оба члена правой части стремятся к нулю при r → ∞. Первый член действительно стремится к нулю при r → ∞, так как при этом e –χ r уменьшается быстрее, чем растет r. Второй же член уравнения не стремится к нулю при r → ∞, поскольку e χr растет быстрее, чем r. Следовательно, он может превратиться в нуль только если a2= 0. Таким образом:
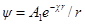
Константу А1определяем из граничного условия, отвечающего r → 0. При r → 0 выражение e – χr стремится к единице и
Но при этом граничном условии потенциал в непосредственной близости от центрального иона определяется только им, а потенциал ионной атмосферы стремится к нулю.
Выражение для потенциала на расстоянии г найдем из следующих соображений. Поскольку потенциал в данной точке это работа удаления W единичного заряда из данной точки в бесконечность, то
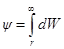
где fK — сила взаимодействия по закону Кулона, равная
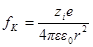
(второй заряд единичный).
Из этих выражений:
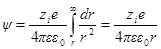
Сравнив данную формулу с выражением ψ = A1 / rполучим:
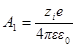 и
и 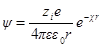
Переписав выражение для потенциала в виде
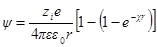
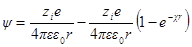
Разложим экспоненту в ряд
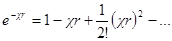
и ограничимся двумя первыми членами. Это ограничение, справед-ливо только для разбавленных растворов, когда χr 3 радиус ионной атмосферы
Таблица 3.2. Расчетные значения радиуса ионной атмосферы 1 / χ
| Тип валентности | Примеры | (1 / χ) ∙ 10 10 (м) при концентрации (кмоль / м 3 ) | |||
| 1,0 | 0,1 | 0,01 | 0,001 | 0,0001 | |
| 1—1 | KCl | 3,0 | 9,6 | 30,5 | 96,4 |
| 1—2; 2—1 | H2SO4, MgCl2 | 1,8 | 5,6 | 19,3 | 55,8 |
| 2—2 | ZnSO4 | 1,5 | 4,8 | 15,3 | 48,2 |
| 1-3; 3-1 | K3Fe(CN)6, LaCl3 | 1,2 | 3,9 | 13,6 | 39,4 |
превышает радиус иона на порядок и больше, то при концентрациях 0,1 кмоль/м 3 и выше радиус ионной атмосферы становится уже соизмеримым с радиусом иона и даже меньше его, что физически невероятно. Правдоподобные значения 1 / χ получаются для водных растворов 1 — 1 -валентных электролитов при концентрациях, меньших 0,01 кмоль/м 3 . Для электролитов с ионами более высоких валентностей этот предел концентраций еще ниже.
Перейдем теперь к расчету собственно межионного взаимодействия. Расчет в теории Дебая и Хюккеля проводится на основе следующего мысленного эксперимента. Допустим, что все ионы в растворе в какой-то момент времени лишены электрических зарядов и с этого момента мы начинаем их заряжать непрерывно, подводя к ним по определенной доле λ их полного заряда zie. В каждый заданный момент времени ионы обладают одинаковой долей λ, их конечного заряда, и потенциал, создаваемый центральным ионом в этот момент, согласно уравнению для потенциала, будет равен:
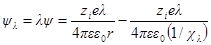
Здесь χλ – радиус ионной атмосферы при заряде иона zieλ,
т. е. χλ = λχ . Следовательно
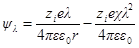
При увеличении заряда иона на dλпроизводится электрическая работа dWэ, равная

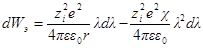
Полная работа заряжения одного иона составит
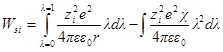
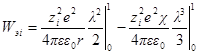
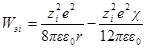
Если в единице объема (1 м 3 ) содержится ni ионов каждого сорта, то электрическая работа заряжения одного сорта ионов будет
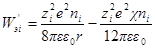
а суммарная электрическая работа заряжения всех сортов ионов в этом объеме составит:
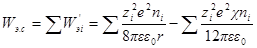
Когда отсутствует ионная атмосфера, т. е. χ = 0, раствор находится в идеальном состоянии, и выражение для общей электрической работы заряжения ионов, находящихся в идеальном состоянии, будет иметь вид:
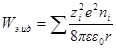
Работа Wэ, расходуемая на заряжение ионной атмосферы, равна Wэ.с – Wэ.ид; для достаточно разбавленных растворов, для которых диэлектрическая проницаемость практически равна диэлектрической проницаемости растворителя, будем иметь:
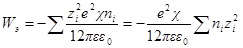
Подставив, как и раньше, вместо ni значение CiN∙10 3 и умно- жив числитель и знаменатель на два, получим:
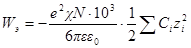
На основании значений электрической работы заряжения ионной атмосферы можно получить выражение для коэффициента активности данного сорта ионов, а затем и среднего ионного коэффициента активности электролита.
разность химических потенциалов μi ид – μi реальн должна равняться изотермической и обратимой работе образования ионной атмосферы, поскольку по теории Дебая — Хюккеля отклонение реального раствора от идеального связано только с электростатическим взаимодействием между ионами. Поэтому
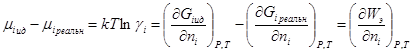
или, учитывая уравнение для электрической работы заряжения
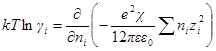
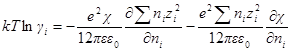
Чтобы решить это уравнение, продифференцируем сперва уравнение характеристической длины по ni
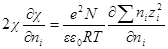
Производная суммы равна zi 2 ,так как все остальные члены суммы от ni не зависят. Следовательно
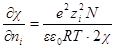
Умножив и разделив правую часть на ∑nizi 2 , можно написать
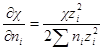
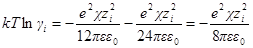
Для логарифма коэффициента активности, таким образом, получаем:
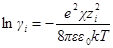
Не следует забывать,что γi в теории Дебая и Хюккеля отличается от γi по Льюису, поскольку в этой теории учитываются только кулоновские силы взаимодействия между ионами и в случае неполной диссоциации соли нужно вводить поправку на долю диссоциированных молекул.
Подставив в выражение для ln γi значение характеристической длины, получим
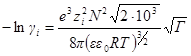
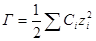
называется ионной силой или ионной крепостью раствора.
Вынесем в уравнении для ln γi универсальные постоянные в общую константу
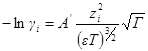
а если ε и T постоянны, то после перехода к десятичным логарифмам имеем:
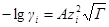
Перейдем теперь к выводу формулы для среднего ионного коэффициента активности. Для этого прологарифмируем уравнение, определяющее γ±:
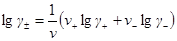
Напишем выражения для γ+ и γ–
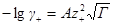 ;
; 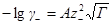
и подставим их в предыдущее уравнение:
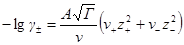
Преобразуем выражение в скобках, помня, что из условия электронейтральности ν+z+ = – ν–z–. Минус здесь получился из-за того, что валентность отрицательного иона ранее была принята отрицательной. Тогда
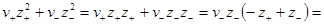
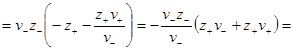
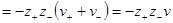
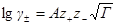
В литературе можно часто встретить эту формулу со знаком минус перед логарифмом среднего ионного коэффициента активности. Это получается в том случае, если при выводе формулы пользуются абсолютными значениями z+ и z–. Тогда
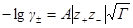
и конечный результат будет тем же.
Полное выражение для среднего ионного коэффициента актив-
ности будет:
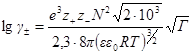
Для водного раствора и стандартной температуры 25 °С после подстановки в это уравнение величин: е= 1,602∙10 –19 Кл, N = 6,022∙10 23 моль –1 ; R = 8,314 Дж/(моль∙К); εε0 = 78,3∙0,885∙10 –11 Ф/м, получим:
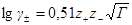
где А =0,51 (м 3 /кмоль) 1/2 .
Это уравнение называется уравнением первого приближения теории Дебая и Хюккеля для коэффициента активности, поскольку оно выведено в наиболее упрощенном приближении к истинной картине взаимодействий в растворе электролита. Можно ожидать, что уравнение будет удовлетворительно описывать поведение растворов электролитов при очень низких концентрациях.
Замечательной особенностью уравнения первого приближения теории Дебая и Хюккеля является отсутствие в нем каких-либо эмпирических постоянных и «подгоночных параметров». Этим уравнением также обосновывается эмпирически установленное ранее правило ионной силы Льюиса и Рендалла, согласно которому:
Во всех очень разбавленных растворах электролитов при равной ионной силе средние ионные коэффициенты активности электролитов одного и того же типа валентности численно одинаковы.
Это правило непосредственно вытекает из формулы первого приближения, поскольку Г = const; z+z– = const и lg γ± = const, ибо все остальные величины, входящие в уравнение Дебая и Хюккеля, являются универсальными постоянными.
В более концентрированных растворах нельзя уже считать ионы геометрическими точками. В связи с этим во втором приближении теории Дебая и Хюккеля вводится предположение, согласно которому ион представляет собой шар радиусом а, в центре которого находится заряд, равный заряду иона. Это предположение физически ближе всего подходит к точечным сольватированным ионам и то, если допустить, что диэлектрическая проницаемость сольват-ного слоя такая же, как и всей массы растворителя.
Радиус а иона является индивидуальной величиной для каждого сорта ионов, и его нельзя определить расчетом или из независимого эксперимента. Фактически это постоянная величина, которую подбирают таким образом, чтобы расчет коэффициента активности по формуле соответствовал бы эксперименту. Кроме того, выше сказано, что во втором приближении теории Дебая и Хюккеля ионы рассматриваются как точечные, окруженные соль-ватной оболочкой и, следовательно, понятие радиуса иона не является вполне строгим.
И, наконец, как это будет ясно из дальнейшего, величина а оказывается одной и той же для всех сортов ионов в данном элек- тролите или в данной смеси электролитов. Таким образом, а — это фактически какая-то средняя величина. Поэтому величину а часто определяют как кажущийся радиус иона или усредненную половину расстояния, на которое максимально могут приблизиться друг, и другу центры ионов.
Уравнение для коэффициента активности выводится аналогии но выводу в первом приближении теории Дебая и Хюккеля, однако определение константы А1сложнее, чем в первом случае, поскольку мы не имеем возможности использовать граничное условие, отвечающее r →0.
Как и раньше, представим себе ион радиуса а, окруженный ионной атмосферой радиусом 1 / χ. Необходимо определить потенциал на поверхности шаровой сферы. Он будет определяться, с одной стороны, зарядом центрального иона и, с другой, — зарядом ионной атмосферы. Потенциал, определяемый зарядом центрального иона, как и раньше, будет ezi / 4πεε0r. Потенциал же ионной атмосферы на расстоянии r от центра обозначим через ψ (r). Тогда
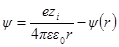 или
или 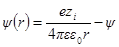
Подставим вместо ψ величину из формулы 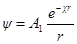 :
:
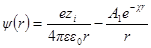
Продифференцируем теперь это уравнение по r: производная первого члена уравнения
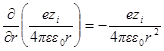
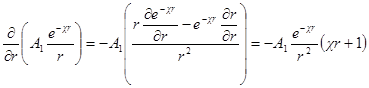
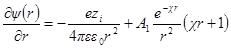
Значения производных первого и второго членов уравнения при r = а должны быть равны друг другу, а это значит, что ∂ ψ (r) / ∂r при r = а должно быть равно нулю. Следовательно, можно получить константу А1в виде:
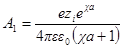
Подставив теперь А1в формулу для ψ (r) при r = а, получим:
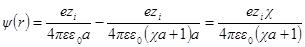
Сравнивая это выражение с аналогичным выражением первого приближения, видим, что учет радиуса иона приводит к отличию величины потенциала ионной атмосферы на выражение 1 / (χa + 1).
Для расчета межиониого взаимодействия проведем, как и раньше, мысленный опыт непрерывного и обратимого заряжения всех ионов от заряда, равного нулю, до заряда, равного полному заряду ezi. В каждый данный момент потенциал ионов будет равен λ-й доле полного потенциала, т. е. ψλ = λψ и χλ = λχ. Тогда:
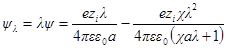
При увеличении заряда на dλпроизводится электрическая работа
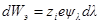
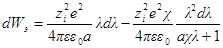
Интегрирование этого уравнения от λ = 0 до λ = 1 дает полную энергию заряжения одного иона. Если ni — число ионов каждого сорта в единице объема, то полная работа образования ионной ат-мосферы составит:
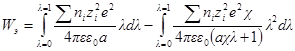
Как и раньше, первый член уравнения дает работу заряжения для идеального случая, а второй — работу образования ионной атмосферы.
Производная Wэпо числу ионов i-гo сорта равняется:
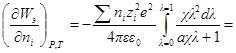
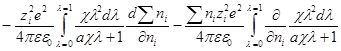
Второй член правой части этого уравнения можно представить в виде:
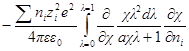
Но ранее показано, что
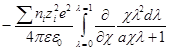
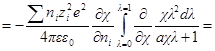
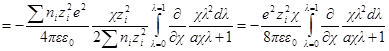
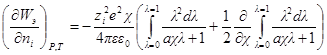
Обозначим: х = aχ; ζ = χaλ, откуда λ = ζ / χa; dx = adχ; dζ = aχdλ. Тогда выражение под интегралом переписывается так:
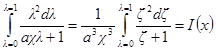
Подставив его в выражение для ∂W / ∂ni, получим:
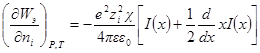
Выражение в квадратных скобках равно 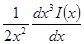 . В самом деле его можно представить в виде:
. В самом деле его можно представить в виде:
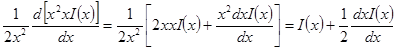 Таким образом
Таким образом
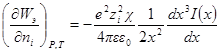
Но как получено ранее
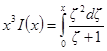
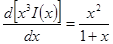 и
и 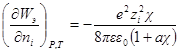
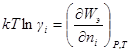
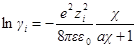
В случае очень разбавленных растворов, когда χ мало и aχ 10 (εТ) –1/2 = = 0,328∙10 10 м –1 (м 3 /кмоль) 1/2 . По физическому смыслу знаменатель в уравнении коэффициента активности учитывает короткодействующие силы между ионами, которые представляются как недеформируемые шары. Величины А и В изменяются при изменении природы растворителя, в то время как а остается приблизительно постоянной.
Значения параметра а определяют из экспериментальных данных по зависимости коэффициента активности от концентрации. Преобразовывая уравнение для коэффициента активности, получим:
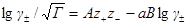
В соответствии с этим уравнением величина lg γ± / 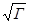 линейно изменяется с lg γ± и из угла наклона прямой определяют параметр а. Ниже приведены значения параметра а для ряда катионов и анионов:
линейно изменяется с lg γ± и из угла наклона прямой определяют параметр а. Ниже приведены значения параметра а для ряда катионов и анионов:
Катионы а, нм Анионы а, нм
H + 0,9 F – , Cl – , Br – , I – , CN – , 0,3
Li + 0,6 NO2 – , NO3 – , OH – , CNS –
Rb + , Cs + , NH4 + , Tl + , Ag + 0,25 CO3 2– , SO3 2– 0,5
Mg 2+ , Be 2+ 0,8 PO4 3– , Fe(CN)6 3– 0,4
Ca 2+ , Cu 2+ , Zn 2+ , Sn 2+ , 0,6
Mn 2+ , Fe 2+ , Ni 2+ , Co 2+
Pb 2+ , Sr 2+ , Ba 2+ , Ra 2+ , 0,5
Fe 3+ , Al 3+ , Cr 3+ , Sc 3+ , 0,9
Y 3+ , La 3+ , In 3+ , Ce 3+
Приведенные значения параметра а позволяют рассчитывать коэффициенты активности электролитов до ионной силы 0,1—0,2. Однако наиболее хорошего совпадения с экспериментальными результатами удается достигнуть, если для каждой концентрации использовать свое значение а. Таким образом, величина параметра оказывается зависящей от концентрации.
Интересно отметить, что по порядку величины произведение (аВ) близко к единице, поэтому для не очень точных расчетов можно применять формулу второго приближения в виде:
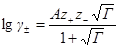
Уравнение второго приближения охватывает зависимость изменения коэффициента активности от концентрации до несколько больших концентраций, чем уравнение первого приближения. Однако оно не может отразить возрастание коэффициента активности многих электролитов с ростом концентрации.
Для охвата единой формулой всей кривой изменения коэффициента активности с концентрацией Хюккель с помощью поправочного члена сГ придал формуле для коэффициента активности вид:
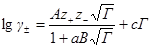
Это уравнение называется уравнением третьего приближения теории Дебая и Хюккеля. Коэффициент с учитывает изменение диэлектрической проницаемости с концентрацией, но он не может быть рассчитан теоретически, подобно коэффициентам А к В. Таким образом, третье приближение Дебая и Хюккеля приводит к формуле, содержащей две эмпирические постоянные, которые подбираются из соображений наилучшего соответствия рассчитанных величин экспериментально определенным.
Шпаргалки к экзаменам и зачётам
студентам и школьникам



Шпаргалки по электрохимии. Часть 2 — Теория электролитов Дебая и Гюккеля
Cмотрите так же.
Теория электролитов Дебая и Гюккеля: ограниченность первого приближения теории, ее причины. Дальнейшее развитие теории.
Сопоставление теории Дебая – Гюккеля с опытом
Таким образом, теория Дебая и Гюккеля позволяет получить такое же уравнение для коэффициента активности, какое было эмпирически найдено для разбавленных растворов электролитов. Из уравнения (1) следует, что коэффициенты активности в растворах с одинаковой ионной силой должны быть одинаковыми. Это согласуется с законом ионной силы Льюиса – Рендалла, который был открыт раньше, чем была создана теория Дебая и Гюккеля. Теория передает зависимость коэффициентов активности в разбавленных растворах от валентного типа электролита и от температуры. Теория, следовательно, находится в качественном согласии с опытом.
Используя выражение для коэффициента активности, можно рассчитать все парциальные термодинамические характеристики раствора (мольную энтропию, мольный объем, теплоемкость, сжимаемость, термическое расширение и т.д.). Теория Дебая и Гюккеля позволила предсказать эффект выделения теплоты при разбавлении растворов электролитов, вызванный тем, что при разбавлении уменьшается взаимодействие ионов. Учитывая, что вышеприведенные уравнения не содержат эмпирических параметров, успехи теории Дебая – Гюккеля следует признать весьма значительными.
При разработке этой теории были сделаны следующие допущения :
1. Число ионов в электролите можно определить из аналитической концентрации электролита, так как он считается полностью диссоциированным ( a = 1). Теорию Дебая и Гюккеля поэтому иногда называют теорией полной диссоциации. Однако ее можно применить и в тех случаях, когда a ¹ 1.
2. Распределение ионов вокруг любого центрального иона подчиняется классической статистике Максвелла – Больцмана.
3. Собственными размерами ионов можно пренебречь по сравнению с расстояниями между ними и с общим объемом раствора. Таким образом, ионы отождествляются с материальными точками, и все их свойства сводятся лишь к величине заряда. Это допущение справедливо только для разбавленных растворов.
4. Взаимодействие между ионами исчерпывается кулоновскими силами. Наложение сил теплового движения приводит к такому распределению ионов в растворе, для которого характерна статистическая шаровая ионная атмосфера. Это допущение справедливо лишь для разбавленных растворов. При повышении концентрации среднее расстояние между ионами уменьшается, и наряду с электростатическими силами появляются другие силы, действующие на более близком расстоянии, в первую очередь, силы Ван-дер-Ваальса. Возникает необходимость учета взаимодействия не только данного иона и его окружения, но и любых двух соседних ионов.
5. При расчетах принимается, что диэлектрические проницаемости раствора и чистого растворителя равны; это справедливо только в случае разбавленных растворов.
Таким образом, три последних допущения Дебая и Гюккеля приводят к тому, что их теория может быть применима только к разбавленным растворам электролитов с ионами низкой валентности . Уравнение (1) соответствует этому предельному случаю и выражает так называемый предельный закон Дебая и Гюккеля или первое приближение теории Дебая и Гюккеля.
Предельный закон Дебая – Гюккеля дает верные значения коэффициентов активности 1 – 1 зарядного электролита, особенно в очень разбавленных растворах (с £ 0,01 моль/л). Сходимость теории с опытом ухудшается по мере увеличения концентрации электролита, увеличения зарядов ионов и уменьшения диэлектрической проницаемости растворителя, то есть с ростом сил взаимодействия ионов.
Дальнейшее развитие теории
Первая попытка усовершенствовать теорию Дебая и Гюккеля и расширить область ее применения была сделана самими авторами. Во втором приближении они отказались от представления об ионах как о материальных точках (допущение 3) и попытались учесть конечные размеры ионов, наделив каждый электролит некоторым средним диаметром а (при этом изменяется и допущение 4). Приписав ионам определенные размеры, Дебай и Гюккель учли тем самым силы некулоновского происхождения, препятствующие сближению ионов на расстояние, меньшее некоторой величины.
Во втором приближении средний коэффициент активности описывается уравнением
lg g ± = —  , (2)
, (2)
где h сохраняет прежнее значение; а условно названо средним эффективным диаметром ионов, имеет размерность длины, фактически — эмпирическая постоянная; В = c /  , В незначительно изменяется с температурой. Для водных растворов произведение В а близко к 1. Формула (2) хорошо описывает поведение многих электролитов вплоть до с = 0,1, однако и во втором приближении нельзя полностью описать весь диапазон зависимости g ± от с. Экспериментальные значения g ± при высоких концентрациях электролита начинают возрастать, и в некоторых растворах это возрастание очень значительно (в водном растворе HClO 4 при m = 16 g ± = 500).
, В незначительно изменяется с температурой. Для водных растворов произведение В а близко к 1. Формула (2) хорошо описывает поведение многих электролитов вплоть до с = 0,1, однако и во втором приближении нельзя полностью описать весь диапазон зависимости g ± от с. Экспериментальные значения g ± при высоких концентрациях электролита начинают возрастать, и в некоторых растворах это возрастание очень значительно (в водном растворе HClO 4 при m = 16 g ± = 500).
Сохранив основные положения второго приближения теории, Гюккель учел уменьшение диэлектрической проницаемости с ростом концентрации растворов. Ее уменьшение вызывается ориентацией диполей растворителя вокруг иона, в результате чего снижается их реакция на эффект внешнего поля. Уравнение Гюккеля выглядит следующим образом:
lg g ± = —  + C I , (3)
+ C I , (3)
где С — эмпирическая константа, лишенная определенного физического смысла. При удачном подборе значений a и С формула Гюккеля хорошо согласуется с опытом и широко используется при расчетах (можно описать экспериментальные данные по g ± до с порядка 1-2). При последовательном уменьшении ионной силы уравнение (3) последовательно переходит в формулу второго приближения теории Дебая и Гюккеля (уравнение (2)), а затем в предельный закон Дебая – Гюккеля (уравнение (1)).
В процессе развития теории Дебая – Гюккеля и последовательного отказа от принятых допущений улучшается сходимость с опытом и расширяется область ее применимости, однако это достигается ценой превращения теоретических уравнений в полуэмпирические.

Рис. 25. Зависимость среднеионного коэффициента активности от ионной силы в водном растворе NaCl :
1 – первое приближение теории Дебая – Гюккеля;
2 – второе приближение теории Дебая – Гюккеля;
3 – третье приближение теории;
4 – экспериментальные данные
Из рис. 25 (кривая 4 – опытная зависимость среднего коэффициента активности от  ) видно, что влияние концентрации на величину коэффициента активности очень значительно; g ± может иметь значение как меньше 1 (вплоть до 0,01), так и много больше 1 (до 500). Это связано с преобладанием того или иного типа взаимодействия частиц: взаимного притяжения либо взаимного отталкивания. Физической основой падения активности по сравнению с концентрацией является взаимное притяжение частиц. Взаимное отталкивание частиц в растворе должно, наоборот, вызывать увеличение активности. В разбавленных растворах электролитов электростатическое притяжение ионов оказывается преобладающим: g ± 1 и падает с ростом концентрации. Учет собственного размера ионов эквивалентен учету сил отталкивания, не позволяющих ионам сблизиться на расстояние, меньшее а. Второе приближение теории, учитывающее этот фактор, приводит к менее резкому уменьшению коэффициента активности (кривая 2) и позволяет описать опытные данные в более широком интервале концентраций. Однако в концентрированных растворах большая часть молекул воды связана ионами, так что добавление новых порций электролита должно сопровождаться разрушением сольватных оболочек и преодолением сил ион-дипольного взаимодействия. Это эквивалентно преобладанию эффекта взаимного отталкивания ионов над их взаимным притяжением; при этом g ± > 1. Таким образом, переход к концентрированным растворам сопровождается резким возрастанием коэффициента активности. Чтобы описать это возрастание, в уравнение третьего приближения теории Дебая и Гюккеля и было формально введено эмпирическое слагаемое C I .
) видно, что влияние концентрации на величину коэффициента активности очень значительно; g ± может иметь значение как меньше 1 (вплоть до 0,01), так и много больше 1 (до 500). Это связано с преобладанием того или иного типа взаимодействия частиц: взаимного притяжения либо взаимного отталкивания. Физической основой падения активности по сравнению с концентрацией является взаимное притяжение частиц. Взаимное отталкивание частиц в растворе должно, наоборот, вызывать увеличение активности. В разбавленных растворах электролитов электростатическое притяжение ионов оказывается преобладающим: g ± 1 и падает с ростом концентрации. Учет собственного размера ионов эквивалентен учету сил отталкивания, не позволяющих ионам сблизиться на расстояние, меньшее а. Второе приближение теории, учитывающее этот фактор, приводит к менее резкому уменьшению коэффициента активности (кривая 2) и позволяет описать опытные данные в более широком интервале концентраций. Однако в концентрированных растворах большая часть молекул воды связана ионами, так что добавление новых порций электролита должно сопровождаться разрушением сольватных оболочек и преодолением сил ион-дипольного взаимодействия. Это эквивалентно преобладанию эффекта взаимного отталкивания ионов над их взаимным притяжением; при этом g ± > 1. Таким образом, переход к концентрированным растворам сопровождается резким возрастанием коэффициента активности. Чтобы описать это возрастание, в уравнение третьего приближения теории Дебая и Гюккеля и было формально введено эмпирическое слагаемое C I .
Следует отметить, что зависимость коэффициента активности в разбавленных растворах электролитов от температуры оказывается весьма незначительной, поскольку повышение температуры сопровождается уменьшением диэлектрической постоянной растворителя, то есть с ростом Т произведение e Т даже несколько уменьшается. В результате при переходе от 0 о к 100 о С коэффициент h изменяется от 0,492 до 0,609; в 0,01 М растворе 1-1 валентного электролита это соответствует уменьшению среднего коэффициента активности всего на 2,7% (от 0,893 до 0,869).
Следует учитывать, что теория дает средний рациональный коэффициент активности ( g ± ), а экспериментальные данные для растворов обычно приводятся в шкале моляльностей ( g ± ¢ ) или молярных концентраций ( f ± ). Средние коэффициенты активности в различных шкалах концентраций связаны следующими соотношениями :
f ± = g ± ¢  , g ± = g ± ¢ (1 + 0,001 n ML m ) , g ± = f ± [ r – 0,001 с ( M – ML ) ] × (1/ r L ) ,
, g ± = g ± ¢ (1 + 0,001 n ML m ) , g ± = f ± [ r – 0,001 с ( M – ML ) ] × (1/ r L ) ,
где r L и ML – плотность и молекулярная масса чистого растворителя соответственно, M – молекулярная масса растворенного вещества, r – плотность раствора. В разбавленных растворах ( с £ 0,01) g ± » g ± ¢ , но при больших концентрациях различие в значениях этих величин становится существенным.
Во всех концентрационных шкалах средние коэффициенты активности при бесконечном разбавлении стремятся к единице, поскольку при этом ион-ионное взаимодействие стремится к нулю и раствор приобретает идеальные свойства. При описании растворов электролитов за стандартное состояние выбирается гипотетический раствор, в котором активности всех ионов равны единице и одновременно отсутствует ион-ионное взаимодействие (естественно, такое стандартное состояние не может быть реализовано).
Существует несколько методов определения активности и коэффициентов активности электролитов. Так, например, активность соли может быть найдена по давлению пара растворителя над раствором, криоскопическим и эбуллиоскопическим методами, по осмотическому давлению. Эти методы для растворов электролитов и неэлектролитов полностью аналогичны. Кроме того, для определения активностей в растворах электролитов может быть использован метод измерения разности потенциалов на концах равновесной электрохимической цепи (метод будет рассмотрен ниже). Во всех методах измеряемые активности в тех или иных координатах экстраполируют на нулевую концентрацию, где g ± ¢ = 1 или f ± = 1.
Активности и коэффициенты активности, полученные различными методами, совпадают в пределах точности эксперимента. Это указывает на то, что термодинамический метод описания взаимодействия в растворах электролитов является правильным и самосогласованным (вспомним, что степени диссоциации, определенные различными методами, оказывались разными).
http://lektsii.org/8-40258.html
http://spargalki.ru/himia/103-shpargalki-electrohimiya.html?start=5