Лекция № 2
План лекции:
ОСНОВНЫЕ ТЕРМОДИНАМИЧЕСКИЕ ПАРАМЕТРЫ ПОВЕРХНОСТНОГО СЛОЯ
ГЕОМЕТРИЧЕСКИЕ ПАРАМЕТРЫ ПОВЕРХНОСТИ
Межфазные поверхности могут существовать только при наличии в системе жидкой или твердой фазы. Межфазную поверхность т-ж определяет профиль поверхности твердого тела. Поверхностный слой на межфазных границах имеет одну часть в первой фазе, другую – во второй.
Удельная поверхность тела определяется отношением площади его поверхности S 1,2 между фазами 1 и 2 к объему тела V .
 (2.1)
(2.1)
Поверхность между фазами может быть отнесена к дисперсной фазе или к дисперсной среде. Обычно при определении удельной поверхности дисперсной системы ее относят к объему дисперсной фазы.
Дисперсные системы с одинаковыми по размерам частицами дисперсной фазы называются монодисперсными, с разными – полидисперсными.
Если поверхность и объем дисперсной фазы междисперсной системы выразить через поверхность и объем отдельных частиц, то число частиц будет входить и в числитель и в знаменатель. Поэтому S уд монодисперсной системы можно определить, зная размер отдельных частиц. Например, для частиц кубических с размерами ребра 1 и сферическим диаметром d :
 (2.2)
(2.2)
для сферических частиц:

 (2.3)
(2.3)
k – коэффициент формы частиц.
Часто S уд относят к массе дисперсной фазы:
 — для квадратных частиц (2.4)
— для квадратных частиц (2.4)
 — для сферических частиц
— для сферических частиц
Изменение S уд с изменением дисперсности зависит от формы частиц.
SHAPE \* MERGEFORMAT

 (2.5)
(2.5)
 (2.5)
(2.5)
 (2.5)
(2.5)
Зависимость возрастает, т. к. возрастает коэффициент формы.
Более конкретная характеристика дисперсности кривизна поверхности, определяемая производной площади поверхности по объему.
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ (  ).
).
 — обусловлено нескомпенсированным полем межмолекулярных сил на межфазной поверхности; — фактор интенсивности поверхностной энергии.
— обусловлено нескомпенсированным полем межмолекулярных сил на межфазной поверхности; — фактор интенсивности поверхностной энергии.
Термодинамическое определение вытекает из объединенного уравнение 1-го и 2-го начал термодинамики. Запишем его для гетерогенной системы относительно изменения внутренней энергии U :
dU = TdS – pdV + dS +  i dni +
i dni +  dq1
dq1
При S, V, n, q — const:
 = (
= (  U / S ) S , U , n , q (2.6)
U / S ) S , U , n , q (2.6)
т.е. — частная производная от внутренней энергии по площади поверхности раздела, при S , V, n, q — const .
 (2.7)
(2.7)
т.е. в общем виде, — частная производная от любого термодинамического потенциала по площади межфазной поверхности при постоянны соответствующих параметрах.
— определяется энергией, приходящейся на единицу площади:
единицы измерения: СИ Дж/м 2 = Н*м/м 2 = Н/м
СГС Эр1/см 2 , Дин/см.
Физический смысл
1. Работа, расходуемая на разрыв межмолекулярной связи. Чем сильней межмокулярные связи в данном теле, тем больше его па границе с газовой фазой, т.е. поверхностное натяжение меньше у неполярных жидкостей со слабыми межмолекулярными связями. Большим поверхностным натяжением обладают вещества с водородными связями (вода).
2. Сила, направленная параллельно к поверхности. Поверхностные молекулы, обладая избыточной энергией, стремятся уйти вглубь конденсированной фазы и сжимают поверхность.
КОГЕЗИОННЫЕ И ПОВЕРХНОСТНЫЕ СИЛЫ
Когезия — взаимодействие молекул, атомов, ионов внутри одной фазы.
W к = 2 (2.8)
т.к. определяется затратой энергии на обратимый разрыв тела по сечению равному единице площади.
Точную информацию о когезии и поверхностном натяжении можно получить из т/д характеристик тел, связанных с парообразованием.
В процессе испарения происходит полный разрыв межмолекулярных связей, поэтому работа когезии определяется энтальпией парообразования.
 H = G п + T S п (2.9)
H = G п + T S п (2.9)
При равновесии при р, Т = const G п = 0 Н = Т S п
Отсюда следует, что, чем больше энтальпия парообразования (т.е. больше работа когезии, а значит и поверхностное натяжение), тем меньше давление насыщенного пара над веществом, т.к.
S п = So — R·ln ( р / ратм ) (2.10),
где So — изменение энтропии парообразования. При температуре кипения.
При кипении р/ратм = 1
L = Ткип So So = L / Ткип (2.11)
L — энтальпия парообразования при Ткип.
Свойства поверхности отражают природу ионов, атомов и молекул в ней.
Для жидкостей и твердых тел когезионные силы выражаются в межмолекулярном взаимодействии, обусловленном ван-дер-ваальсовыми и водородными связями. Оно отличается от химического взаимодействия отсутствующей специфичностью и насыщаемостью небольшими энергиями. Разрыв таких связей приводит к формированию поверхностей, способных образовывать ван-дер-ваальсовые и водородные связи с молекулами, попадающими на эту поверхность.
ВНУТРЕННЯЯ (ПОЛНАЯ) УДЕЛЬНАЯ ПОВЕРХНОСТНАЯ ЭНЕРГИЯ. ЗАВИСИМОСТЬ ЭНЕРГЕТИЧЕСКИХ ПАРАМЕТРОВ ПОВЕРХНОСТИ ОТ ТЕМПЕРАТУРЫ.
За толщину поверхностного слоя принимают расстояние по обе стороны от границы раздела фаз, за пределами которой свойства слоя перестают отличаться от свойств объемных фаз.
Установление границ поверхностного слоя со стороны объемных фаз трудная задача. Гиббс предложил относить все изменения т/д параметров в слое к разделяющей поверхности, не имеющей объема или толщины (метод избыточных величин Гиббса).
G = G 1 + G 2 + * S (2.12)
G – энергия Гиббса системы;
G 1 и G 2 – энергия Гиббса объемных фаз;
S – поверхностная энергия Гиббса.
Избыточная внутренняя энергия:
Индекс « s » указывает на то, что потенциалы отнесены к единице поверхности.
q s = TS s теплота образования единицы поверхности (скрытая теплота образования, она равна количеству теплоты, которую нужно сообщить телу, чтобы при постоянной температуре увеличить его поверхность на единицу площади).
Учитывая, что G s = , можно написать:
U s = + qs (2.15)
Из уравнения 2.15 следует, что внутренняя энергия поверхности складывается из энергии Гиббса и теплоты образования поверхности.
Из объединенного уравнения, при постоянстве всех параметров, кроме температуры, имеем:
dGs = — S dT или (  Gs/ T) p = -S = -q s /T (2.16)
Gs/ T) p = -S = -q s /T (2.16)
Подставляя (2.16) в (2.14) или (2.15), получаем:
U s = — T ( / T ) p (2.17)
(2.17) – уравнение Гиббса-Гельмгольца, оно связывает поверхностную энергию с поверхностным напряжением, следовательно, для определения полной поверхностной энергии нужно знать зависимость поверхностного напряжения от температуры. Конкретную зависимость можно получить только экспериментально, но качественные выводы можно сделать из уравнения (2.17).
qS – всегда больше 0. (для индивидуального вещества).
( G s / T ) p = ( / T ) p 0, следовательно, уменьшается с увеличением температуры. Для большинства неполярных жидкостей эта зависимость линейна и в первом приближении ее можно записать, как:
T = 0 — a D T (2.18)
 — постоянная, равная температурному коэффициенту с обратным знаком.
— постоянная, равная температурному коэффициенту с обратным знаком.
Для большинства жидкостей поверхностная энергия почти не зависит от температуры.
Чтобы убедиться в этом проинтегрируем по температуре (2.17)
( U s / T) = ( / T) – ( / T) – T( 2 / T 2 )
( U s / T) = -T( 2 / T 2 )
— уменьшается с увеличением температуры, то  (см. 2.18), таким образом,
(см. 2.18), таким образом,
( Us / T ) = 0, что означает независимость US от температуры.
Природа поверхностной энергии. Поверхностное натяжение
» data-shape=»round» data-use-links data-color-scheme=»normal» data-direction=»horizontal» data-services=»messenger,vkontakte,facebook,odnoklassniki,telegram,twitter,viber,whatsapp,moimir,lj,blogger»>
Природа поверхностной энергии. Поверхностное натяжение. ТЕРМОДИНАМИКА МЕЖФАЗНОЙ ПОВЕРХНОСТНОСТИ
Наиболее важной характеристикой поверхности является поверхностное натяжение а.
Поверхностное натяжение – это избыточная энергия, приходящаяся на единицу площади поверхности:

Физическая природа поверхностного натяжения в нескомпенсированности поля межмолекулярных сил на межфазных поверхностях.
Поверхностное натяжение характеризует различия в интенсивности межмолекулярных взаимодействий граничащих фаз. Чем сильнее межмолекулярные связи в веществе, тем больше поверхностное натяжение на его межфазной поверхности.
С термодинамической точки зрения, поверхностное натяжение определяется частной производной от любого термодинамического потенциала по величине площади межфазной поверхности при постоянстве других параметров. Используя потенциал Гиббса (G), можно записать

где р – давление; Т- температура; п. – число молей компонентов.
Поверхностная энергия является частью свободной энергии системы в целом. В самопроизвольных процессах эта энергия может быть снижена либо за счет уменьшения площади поверхности и изменения ее формы, либо за счет снижения поверхностного натяжения.
Факторы, влияющие на поверхностное натяжение
Поверхностное натяжение на границе раздела фаз между жидкостью и газом определяют следующие факторы: химическая природа вещества, температура, природа граничащих фаз, наличие примесей, заряд поверхности, кривизна поверхности жидкости.
Энергия межмолекулярных взаимодействий зависит от температуры, а значит, имеет выраженную температурную зависимость.
Взаимосвязь поверхностного натяжения, температуры и полной поверхностной энергии (внутренней энергии поверхностного слоя) Us выражается уравнением Гиббса – Гельмгольца:

Для многих веществ температурные коэффициенты поверхностного натяжения находятся в диапазоне от -0,1 до 0,2 мДж/(м 2 *К).

Рис. 2.1. Зависимость поверхностного натяжения жидкостей от температуры: вода (У), глицерин (2), нитробензол (3), гексан (4).
С повышением температуры поверхностное натяжение уменьшается, а теплота образования единицы площади поверхности увеличивается. Это объясняется тем, что с повышением температуры расстояние между молекулами в жидких телах увеличивается, и соответственно равнодействующая межмолекулярных сил (и, следовательно, поверхностное натяжение) уменьшается. С помощью температурного коэффициента можно определить поверхностное натяжение при любой температуре, если известно значение поверхностного натяжения при какой-то температуре, т. е.

Появление кривизны поверхности из-за стремления системы
к минимуму поверхностной энергии ведет не только к изменению площади межфазной поверхности, но и к появлению избыточного давления внутри фаз. Взаимосвязь между избыточным внутренним давлением в теле и кривизной его поверхности выражается уравнением Лапласа:

Искривление поверхности вызывает повышение или понижение давления в фазе по сравнению с плоской поверхностью фазы такого же химического состава. Очевидно, что это приводит к изменению термодинамических параметров вещества, которые определяют его физические свойства и реакционную способность. Понятие термодинамическая реакционная способность вещества характеризует его способность изменять химический или фазо
вый состав, т. е. вступать в химическую реакцию или переходить в новую фазу (например, испаряться или конденсироваться, растворяться).
У тел с искривленной поверхностью меняется не только внутреннее давление, но и его свободная энергия

Уравнение показывает, что приращение реакционной способности пропорционально 1/r- кривизне поверхности или дисперсности. Чем выше кривизна поверхности или дисперсность, тем выше ее влияние на реакционную способность.
При переходе из газообразного состояния в жидкое

гдер – давление пара над искривленной поверхностью; ps– давление пара над плоской поверхностью.
Тогда уравнение, записанное для сферической поверхности, называется уравнением капиллярной конденсации Кельвина (Томсона):

Из анализа данного уравнения можно сделать вывод о том, что при положительной кривизне жидкости (капля в невесомости или на поверхности твердого тела при отсутствии или неполной смачиваемости) над ней создается повышенное по сравнению с плоской поверхностью давление пара, т. е. испаряется больше жидкости. При отрицательной кривизне (жидкость, смачивающая капилляр) количество испарившейся жидкости в равновесии с ее паром будет меньше по сравнению с плоской поверхностью; иными словами, конденсация будет происходить при меньшем давлении паров.
При появлении кривизны поверхности и увеличении степени дисперсности уменьшается и температура фазовых переходов.
Свободная энергия единицы поверхности и поверхностное натяжение
3.1. Поверхностные свойства веществ
Если взять частицу внутри жидкости и, рассматривая ее как точку, провести сферу радиуса действия межчастичных сил, то за промежуток времени, больший по сравнению с периодом собственных колебаний, на частицу будут действовать силы, одинаковые во всех направлениях.

Следовательно, силовое поле атомов или молекул в объеме жидкости симметрично. Частицы в глубине и на поверхности имеют разные силовые поля. На поверхности жидкости частица будет испытывать преимущественное притяжение со стороны жидкой фазы. Если вторая фаза — пар или газ, то силами взаимодействия со стороны другой фазы можно пренебречь. Если вторая фаза — жидкость, то равнодействующая будет другая.

В общем случае межчастичные силы на межфазной границе несимметричны. Наличие такой асимметрии силового поля приводит к тому, что появляется равнодействующая, направленная перпендикулярно поверхности. Под действием этой силы поля частица втягивается вглубь жидкости. Если частицы уподобить шарикам, то перемещение подвижных частиц жидкости можно проиллюстрировать следующим рис. 3.3:

Когда частица из поверхностного слоя уйдет в объем жидкости, между оставшимися двумя соседними частицами будут действовать силы притяжения. Силы притяжения между частицами В и С сближают поверхностные частицы, поэтому жидкость самопроизвольно сокращает свою поверхность. Такой характер поведения жидкости обусловлен ее текучестью. Если на жидкость не действуют никакие другие силы, то жидкость принимает форму сферы, обладающей минимальной поверхностью. Если же действуют силы тяжести, то форма жидкости может быть другой. При малом объеме жидкости поверхностные силы намного превосходят силы тяжести и жидкость собирается в сферическую капельку. По мере увеличения объема жидкости эти силы становятся соизмеримыми и получается приплюснутая капля. В случае большого объема жидкости силы тяжести значительно больше поверхностных сил и жидкость принимает форму сосуда.

Если увеличивать поверхность жидкости, то на это увеличение нужно затрачивать работу: δА — работа увеличения поверхности на dω. При обратимом изотермическом процессе эта работа максимальна и равна убыли свободной энергии системы: δАмакс = –dF. Изменение свободной энергии, отнесенной к единице поверхности при постоянных объеме и температуре (V, T = const), называется свободной энергией единицы поверхности, или поверхностным натяжением.

F Работа δА отрицательна, так как работу совершаем мы, а не система. Размерность поверхностного натяжения:

Энергия на единицу поверхности. Эти размерности можно преобразовать:

Сила, действующая на поверхности жидкости, направленная по касательной к этой поверхности. Эту силу называют поверхностным натяжением — это сила на единицу длины, действующая по касательной к поверхности. Количественно свободная энергия единицы поверхности жидкости равна поверхностному натяжению, однако отождествлять их нельзя. Энергия — это скалярная величина, а сила — векторная. Они совпадают только в изотропных средах. В анизотропных средах (кристаллах) эти характеристики могут существенно отличаться. В кристаллических телах сила — не вектор, а тензор. Вектор можно задать тремя числами, а тензор определяется числовой матрицей, т. е. набором чисел. Если этих чисел три, то имеем вектор r (x, y, z). Если состояние напряженное, то надо знать напряжение по различным направлениям.
Натяжение в общем случае нужно рассматривать как тензор. Поэтому в кристаллах нельзя отождествлять свободную энергию и поверхностное натяжение. В жидкости они совпадают количественно, и обычно в литературе их отождествляют:

Поверхностное натяжение жидкостей обычно определяют на границе с их собственным насыщенным паром или инертным газом. Величина свободной энергии зависит от свойств жидкости и является вполне определенной. Поверхностное натяжение жидкости — ее свойство, ее характеристика при данной температуре. Натяжение на границе двух конденсированных фаз (ж1‑ж2, ж‑тв) зависит от свойств каждой жидкости и является их совместной характеристикой. Поэтому его называют межфазным натяжением. Размерность та же.

Для конденсированных фаз при малых давлениях F ≈ G, и обычно технологи пользуются энергией Гиббса. F (V, T), G (P, T) — в скобках записаны независимые переменные.

Правильнее было бы обозначать как σ V и σ Р . σ V — работа образования единицы поверхности при неизменном объеме системы; σ Р — работа образования единицы поверхности при постоянном давлении. Если изменение объема ΔV мало, что характерно для конденсированных фаз, то этими различиями можно пренебречь.
Для чистых жидкостей при малых давлениях PdV → 0 . Если давление большое, то σР > σV на работу против сил внешнего давления. Для чистых жидкостей это справедливо, а для растворов эти понятия не тождественны (т. к. имеем различные структурные коэффициенты). В дальнейшем мы будем рассматривать небольшие давления и считать, что σР = σV
ПРИМЕРЫ РЕШЕНИЯ ЗАДАЧ
Пример 2.1.
При конденсации тумана, состоящего из капель кадмия, образовалось 12,5 • 10 -6 м 3 жидкого кадмия. Поверхностное натяжение при температуре конденсации равно 570 мДж/м 2 . Свободная поверхностная энергия всех капель составляла 53 Дж. Вычислите дисперсность и диаметр капель жидкого кадмия.

Пример 2.2.
Рассчитать давление насыщенных паров над каплями воды с дисперсностью 0,1 нм -1 при 293 К. Давление над плоской поверхностью при этой температуре составляет 2338 Па, р = 1 г/см 3 , поверхностное натяжение 72,7 мДж/м 2 , мольный объем 18 • 10 -6 м 3 /моль.

Пример 2.3.
В воздухе, содержащем пары воды, образуется туман при температуре 270 К. Степень пересыщения составляет 3,01. Поверхностное натяжение 73 мДж/м 2 , мольный объем вещества в конденсированном состоянии 18*10 -6 м 3 /моль. Рассчитать критический размер ядер конденсации и число молекул, содержащихся в них.

Пример 2.4.
По экспериментальным данным (табл. 2.1) температурной зависимости поверхностного натяжения найти температурный коэффициент.
Внутренняя (полная) удельная поверхностная энергия
При образовании единицы поверхности S изменяется внутренняя поверхностная энергия.
Вернемся к объединенному уравнению первого и второго начал химической термодинамики для этого случая:
где S – энтропия, μi – химический потенциал компонента, φ – потенциал поверхности.
При неизменном объеме системы V, постоянном составе вещества n, отсутствии заряда q на поверхности (V, n, q = const) получаем:
Проинтегрируем полученное уравнение от s до (s+1) (при образовании единичнгой поверхности):
 , (2.29)
, (2.29)
где σ = GS, а TSS = qs– скрытая теплота образования единицы поверхности, величина qs всегда >0.
Внутренняя поверхностная энергия единицы поверхности больше поверхностной энергии Гиббса (*) на теплоту образования единицы поверхности. Поэтому ее обычно называют полной поверхностной энергией.
Запишем уравнение для изобарно-изотермического потенциала в дифференциальной форме:
где S – энтропия системы; Т – температура, V – объем, P – давление.
Из этого уравнения следует, что температурная зависимость энергии Гиббса:
 ; а т.к. σ = GS, то и
; а т.к. σ = GS, то и  (2.32)
(2.32)
Отсюда qs = TS=  (2.33)
(2.33)
Для конденсированных систем из-за очень небольшой разницы в объемах полная внутренняя энергия U и энтальпия Н практически совпадают, поэтому уравнение Гиббса-Гельмгольца (**) , связывающее полную поверхностную энергию или энтальпию с энергией Гиббса в этом случае можно записать:
 . (2.34)
. (2.34)
Как следует из уравнения (2.34), для определения Us или Нs надо знать σ и зависимость σ = f(Т). Конкретную зависимость можно определить только экспериментально.
Значения Us, Нs, qs для некоторых веществ приведены в табл. 2.3.
Термодинамические характеристики поверхности некоторых жидкостей на границе их с воздухом
| Жидкость | -dσ/dТ 10 3 , Дж/м 2 К | σ 10 3 ,Дж/м 2 | Us,10 3 ,Дж/м 2 | qs 10 3 ,Дж/м 2 |
| Гексан (С6Н8) Этанол (С2Н5ОН) Уксусная кислота (СН3СООН) Вода (Н2О) Глицерин (С3Н2О3) Бензол (С6Н6) Ртуть (Нg) | 0,11 0,1 0,1 0,16 0,05 0,13 0,233 | 18,41 22,03 27,79 72,7 59,2 28,9 473,5 | 49,5 46,4 55,9 119,0 74,25 67,0 542,0 | 31,09 24,37 28,11 46,15 15,05 31,72 68,5 |
Значительный вклад в значения Us и Нs, вносит теплота qs – энтропийная составляющая. Это объясняется тем, что при переходе молекул и атомов из объема тела на поверхность связи разрываются и на поверхности вещество оказывается в состоянии, близком к паровой фазе.
Для большинства жидкостей, особенно неполярных, Us и Нs не зависят от температуры. Этот вывод следует и из уравнения (2.34)
Убедимся в этом. Продифференцируем по Т уравнение Гиббса-Гельмгольца:
 (2.35)
(2.35)
а т.к. 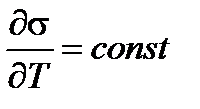 , то
, то  , отсюда
, отсюда  . (2.36)
. (2.36)
Поэтому при увеличении температуры поверхностное натяжение уменьшается, Us не зависит от температуры, а qs возрастает.
Температурные зависимости для систем ж/г показаны на рис. 2.11:
Рис.2.11. Температурная зависимость термодинамических параметров в поверхностном слое: свободной энергии σ, полной энергии Us, скрытой теплоты образования поверхности qs
Адсорбция
2.4.1. Основные понятия и определения
Адсорбция – процесс самопроизвольного изменения концентрации (перераспределения) компонентов системы между поверхностным слоем и объемной фазой.
Более плотная фаза называется адсорбентом (в жидком или твердом агрегатном состоянии).
Вещество, которое адсорбируется, называется адсорбат или адсорбтив.
Обратный процесс называется десорбцией.
Для количественного описания адсорбции используются две величины:
1. Абсолютная адсорбция А – число моль или г адсорбата, приходящееся на единицу поверхности или массы адсорбента.
Единицами измерения А являются моль/м 2 , моль/г или моль/см 3 . Экспериментально А определяют весовым методом (например, на весах Мак-Бена) при изучении адсорбции из газовой фазы на твёрдом адсорбенте. Увеличение массы (пересчитанное в моль) адсорбента, подвешенного на весах, равно именно А.
2. Избыточная адсорбция (гиббсова) Г– избыток адсорбата в поверхностном слое по сравнению с его количеством в таком же объеме фазы, приходящийся на единицу поверхности или массы адсорбента.
Измеряют избыточную адсорбцию также в моль/м 2 , моль/г или моль/см 3 . Экспериментально Г определяют по разности концентраций адсорбата в растворе до и после адсорбции (как это делается в лабораторном практикуме).

По своему физическому смыслу А всегда положительна (А > 0). Значение же Г может быть как положительным (вещество концентрируется на поверхности), так и отрицательным (вещество избегает поверхности, как в случае адсорбции ПИВ).
По определению А всегда больше Г, но при малых концентрациях адсорбата (можно пренебречь количеством вещества в слое фазы по сравнению с количеством его у поверхности) и сильной его адсорбции А » Г. Обычно это наблюдается в водных растворах ПАВ.
Установлен ряд приближенных критериев, совокупность которых позволяет на основании экспериментальных данных различить физическую и химическую адсорбции.
1. Физическая адсорбция происходит под влиянием сил Ван-дер-Ваальса и по своей природе аналогична процессам конденсации паров адсорбата. Поэтому теплота её близка к теплотам конденсации и составляет –(5 – 40) кДж/моль. Теплота хемосорбции соизмерима с теплотами химических реакций и составляет обычно –(80 – 400) кДж/моль.
Однако хемосорбция из жидких растворов может сопровождаться выделением теплоты, близкой к теплоте физической адсорбции. Таким образом, если наблюдаемые теплоты адсорбции превышают -80 кДж/моль, то можно с достаточной уверенностью утверждать, что исследуемое явление — хемосорбция. Нельзя, однако, делать вывод о физической природе адсорбции в случае малой величины её теплоты.
2. Температурная область протекания физической адсорбции не может значительно превышать температуру кипения адсорбата при давлении опыта. Так, при атмосферном давлении физическая адсорбция воды ограничена Т≈ 100 0 С. Хемосорбция же может происходить как при низких, так и при гораздо более высоких температурах.
3. Физическая адсорбция на непористых адсорбентах протекает практически мгновенно, и скорость её слабо зависит от температуры. Хемосорбция, как и любая химическая реакция, протекает через образование активированного комплекса с преодолением энергии активации, т.е. является активированной адсорбцией. Скорость такой адсорбции сильно зависит от температуры (эта зависимость передается уравнением Аррениуса (*) ).
Однако бывают случаи, например, при хемосорбции кислорода и водорода на поверхности металлов, когда адсорбция протекает очень быстро и практически без зависимости её скорости от температуры.
4. Однозначным критерием установления природы адсорбции является отсутствие значительной температурной зависимости скорости десорбции.
Энергия активации десорбции равна сумме энергии активации адсорбции и теплоты адсорбции. Слабая зависимость скорости десорбции от температуры возможна лишь при малых величинах как энергии активации, так и теплоты адсорбции, а это характерно лишь для физической адсорбции.
5. Физическая адсорбция не специфична: она происходит на любых поверхностях (если температура опыта ниже температуры кипения адсорбата).
Благодаря этой особенности физическая адсорбция и может быть использована для измерения общей поверхности твердых тел. В противоположность этому хемосорбция происходит только на тех адсорбентах, с поверхностями которых возможно химическое взаимодействие адсорбата (между ними имеется химическое сродство).
6. Физическая адсорбция может приводить к образованию полимолекулярных пленок (полимолекулярной адсорбции), так как силы взаимодействия в последующих слоях практически не отличаются от сил взаимодействия в первом слое. При хемосорбции химическое взаимодействие требует непосредственного контакта адсорбата с поверхностью и возможность полимолекулярной адсорбции исключается.
Однако количество адсорбированного вещества при хемосорбции может в некоторых случаях превышать однослойное покрытие вследствие проникновения адсорбата на некоторую глубину приповерхностного слоя в междоузлия кристаллической решетки адсорбента. При хемосорбции кислорода на серебре или платине адсорбированное количество может более, чем в 3 раза превышать число атомов кислорода, отвечающее монослойному покрытию поверхности. При этом не образуется объемная фаза оксида.
7.Химическая адсорбция локализована, т.е. на каждом центре адсорбции поверхности может адсорбироваться только одна молекула адсорбата (поверхность можно представить шахматной доской, на каждой клетке которой может находиться только одна фигура). Физическая же адсорбция нелокализована, т.е. в этом случае нет жесткой связи молекул адсорбата и центров адсорбции.
Приведенные критерии, рассматриваемые в отдельности, не всегда позволяют однозначно охарактеризовать тип адсорбции, но примененные совместно обычно позволяют надежно отличать физическую адсорбцию от хемосорбции.
Надо, однако, иметь в виду, что при отсутствии резкой границы между явлениями физического и химического взаимодействия возможна адсорбция, характеризующаяся промежуточными свойствами между физической адсорбцией и хемосорбцией.
Часто в литературе можно встретить утверждение, что физическая адсорбция – обратимая, а хемосорбция – необратимая. Оно не корректно: хемосорбция, как и любая химическая реакция, идёт до установления равновесия, когда скорость адсорбции равна скорости десорбции. Термин «необратимая адсорбция» следует использовать лишь в тех случаях, когда химическая природа адсорбирующихся и десорбирующихся молекул различна (молекулы распадаются на фрагменты и при десорбции с поверхности выделяются совсем другие частицы). Так, при десорбции хемосорбированного на платине бензола с поверхности удаляется целый набор углеводородов – от метана до циклогексана.
В общем случае адсорбция является функцией давления Р (для газов) или концентрации С (для жидких растворов) и температуры, т.е. изображается на плоскости в координатах а= f(P,T) или Г = f(C,T).
Обычно один из параметров поддерживают постоянным и адсорбцию графически изображают в виде следующих кривых (рис.2.12):
1. Изотерма— это зависимость адсорбции от давления газа или от концентрации раствора при постоянной температуре.
2. Изобара— это зависимость адсорбции от температуры при постоянном давлении газа (изопикна— при постоянной концентрации).
3. Изостера— зависимость давления (или концентрации) от температуры при постоянной адсорбции.
На практике для графического изображения адсорбции чаще всего используют изотермы.

Рис.2.12. Зависимости адсорбции а от различных параметров
http://farmf.ru/lekcii/priroda-poverhnostnoj-energii-poverhnostnoe-natyazhenie/
http://lektsia.com/2x3a89.html