Осмос. Осмотическое давление. Закон Вант-Гоффа. Зависит ли осмотическое давление от природы растворённого вещества?
Осмосом называют преимущественно одностороннее проникновение молекул растворителя(диффузию) через полунепроницаемую мембрану из растворителя в раствор или из раствора с меньшей концентрацией в раствор с большей концентрацией.
Осмотическим давлением называют величину, измеряемую минимальным гидравлическим давлением, которое нужно приложить к раствору, чтобы осмос прекратился.
Осмотическое давление разбавленных растворов неэлектролитов прямо пропорционально молярной концентрации, коэффиценту пропорциональности и абсолютной температуре: π =С(Х)RT,
Где π- осмотич давление,кПа; С(Х)- молярная концентрация, моль/л
С(Х)= n/V, где n-число молей неэлектролита, V- объём раствора; R- универсальная газовая постоянная,равная 8,31кПа*л/(моль*К); T-абсолютная температура, K.
π = n/V * RT или πV= nRT.
Чтобы согласовать количественное описание коллигативных свойств электролитов и законы идеальных растворов, Вант-Гофф ввёл поправочный коэффицент i, называемый изотоническим коэффицентом
Осмотическое давление зависит от концентрации растворенного вещества и температуры. Так, при увеличении концентрации с сахарозы в воде в два раза осмотическое давление возрастает примерно в два раза, при увеличении концентрации c в три раза осмотическое давление возрастает почти во столько же и т. д.
28. Гипо-, гипер- и изотонические растворы. Изотонический коэффицент
Изотонический раствор- жидкость внутренней среды или искусственно приготовленный раствор имеет такое же осмотическое давление, как нормальная плазма крови, подобную жидкую среду или раствор
Гипертонический раствор жидкость с более высоким осмотическим давлением
Гипотонический раствор жидкость с более низким осмотическим давлением
Чтобы согласовать количественное описание коллигативных свойств электролитов и законы идеальных растворов, Вант-Гофф ввёл поправочный коэффицент i, называемый изотоническим коэффицентом
Изотонический коэффициент или коэффициент Вант-Гоффа (i)– это отношение суммы числа ионов и непродиссоциировавших молекул электролита к начальному числу молекул электролита, по его величине вычисляют степень электролитической диссоциации:
Формулы для расчета коллигативных свойств разбавленных растворов электрполитов с учетом изотонического коэффициента имеют вид:
— осмотическое давление Росм = ί См RT;
— 1-й закон Рауля ΔР / Р(z) = ί Сm ;
Нетрудно увидеть, что изотонический коэффициент ί может быть вычислен как отношение ΔР, Δtкp, Δtкип, Росм, найденных экспериментально на опыте, к тем же величинам, вычисленным без учета диссоциации электролита (Δрвыч, Δtкp выч, Δtкип выч; Росм выч):
| 29. Роль осмоса в биологических системах. Плазмолис и лизис. Осмотическое давление обеспечивает переход растворителя через полунепроницаемую мембрану от р-ра менее концентрированного к р-ру более концентрированному, поэтому оно играет важную роль в распределении воды между внутренней средой и клетками организма. Лизис-набухание клеток, разрыв оболочек, вытекание клеточного содержимого, вследствии помещения клеток в гипотонический р-р. Плазмолис-сморщивание клеток, при их помещении в гипертонический р-р. | |
| 30. Что называют буферными растворами? Буферные р-ры-это р-ры рН которых меняется незначительно при разбавлении или при добавлении небольших количеств китслоты или щелочи. | . |
Изотонический раствор- жидкость внутренней среды или искусственно приготовленный раствор имеет такое же осмотическое давление, как нормальная плазма крови, подобную жидкую среду или раствор
Гипертонический раствор жидкость с более высоким осмотическим давлением
Гипотонический раствор жидкость с более низким осмотическим давлением
Чтобы согласовать количественное описание коллигативных свойств электролитов и законы идеальных растворов, Вант-Гофф ввёл поправочный коэффицент i, называемый изотоническим коэффицентом
Изотонический коэффициент или коэффициент Вант-Гоффа (i)– это отношение суммы числа ионов и непродиссоциировавших молекул электролита к начальному числу молекул электролита, по его величине вычисляют степень электролитической диссоциации:
Формулы для расчета коллигативных свойств разбавленных растворов электрполитов с учетом изотонического коэффициента имеют вид:
— осмотическое давление Росм = ί См RT;
— 1-й закон Рауля ΔР / Р(z) = ί Сm ;
Нетрудно увидеть, что изотонический коэффициент ί может быть вычислен как отношение ΔР, Δtкp, Δtкип, Росм, найденных экспериментально на опыте, к тем же величинам, вычисленным без учета диссоциации электролита (Δрвыч, Δtкp выч, Δtкип выч; Росм выч):
36. Объясните, почему большинство буферных систем организма имеет буферную емкость по кислоте больше, чем по основанию.
Потому что в живом организме в результате метаболизма образуются большие количества кислых продуктов. Так, в организме человека за сутки образуется такое количество различных кислот, которое эквивалентно 20-30 л однонормальной сильной кислоты. И для того чтобы поддерживать рH организма, у буферных систем организма буферная емкость по кислоте больше, чем по основанию.
37. Патологические явления: ацидоз и алкалоз
Алкалоз — одна из форм нарушения кислотно- щелочного равновесия организма; характеризуется абсолютным или относительным избытком оснований, т.е. Веществ, присоединяющих ионы водорода(протоны), по отношению к кислотам, отщепляющим их. Алкалоз может быть компенсированным или некомпенсированным в зависимости от значения pH. При компенсированном алкалозе pH крови удерживается в пределах нормальных величин (7,35-7,45), отмечаются лишь сдвиги в буферных системах и физиологических регуляторных механизмах. При некомпенсированном алкалозе pH превышает 7,45, что обычно связано со значительным избытком оснований инедостаточностью физико-химических и физиологических механизмов регуляции кислотно- щелочного равновесия. Ацидоз — сдвиг кислотно-щелочного равновесия в организме в сторону относительного увеличения количества анионов кислот, характеризуется абсолютным или относительным избытком кислот, т.е. веществ, отдающих ионы водорода (протоны), по отношению к основаниям, присоединяющим их. Ацидоз также может быть компенсированным или некомпенсированным в зависимости от значения pH. При компенсированном ацидозе pH крови смещается к нижней границе физиологической нормы (7,35). При более выраженном сдвиге в кислую сторону (pH менее 7,35) ацидоз считается некомпенсированным. Такой сдвиг обусловлен значительным избытком кислот и недостаточностью физико-химических и физиологических механизмов регуляции кислотно- щелочного равновесия.
38. Какое химическое равновесие поддерживают в организме буферные системы?
В организме буферные системы поддерживают кислотно-щелочное равновесие. В организме человека особенно большую роль играют белковый, гидрокарбонатный, гемоглобиновый и фосфатный буферы.
39. Какая буферная система вносит максимальный относительный вклад в поддержание протолитического гомеостаза во внутренней среде эритроцитов? Максимальный относительный вклад в поддержание протолитического гомеостаза во внутренней среде эритроцитов вносит гемоглобиновая буферная система.
40.Какие соединения называются координационными? Приведите примеры.
Комплексные соединения или координационные соединения — частицы (нейтральные молекулы или ионы), которые образуются в результате присоединения к данному иону (или атому), называемому комплексообразователем, нейтральных молекул или других ионов, называемых лигандами. Примеры: [Cu(NH3)4] SO4 — сульфат тетраамминмеди (II) K4[Fe(CN)6] — Гексацианоферра́т(II) ка́лия [Pt(NH3)2Cl2] — транс-Дихлородиамминплатина(II)
41. Классификация координационных соединений
Координационные соединения классифицируют: Катионные комплексы образованы вñ1)По заряду комплекса: результате координации вокруг положительного иона нейтральных молекул (H2O, NH3и др.). [Zn(NH3)4]Cl2 — хлорид тетраамминцинка(II) [Co(NH3)6]Cl2 — хлорид гексаамминкобальта(II) Анионные комплексы: в ролиñ комплексообразователя выступает атом с положительной степенью окисления, а лигандами являются простые или сложные анионы. K2[BeF4] — тетрафторобериллат(II) калия Li[AlH4] — тетрагидридоалюминат(III) лития K3[Fe(CN)6] — гексацианоферрат (III) калия Нейтральные комплексы образуются приñ координации молекул вокруг нейтрального атома, а также при одновременной координации вокруг положительного иона — комплексообразователя отрицательных ионов и молекул. [Ni(CO)4] — тетракарбонилникель [Pt(NH3)2Cl2] — дихлородиамминплатина(II) 2) По числу мест, занимаемых лигандами в координационной сфере Монодентатные лиганды.ñ Полидентатные лиганды.ñБидентатные лиганды.ñ
42. Природа химической связи в комплексных соединениях
Во внутренней сфере комплексного соединения связь между комплексообразователем и лигандами ковалентная, образованная по донорно-акцепторному механизму. Ион или атом- комплексообразователь является акцептором, а лиганды являются донорами электронных пар.
№43) Как расчитывается общая и стуаенчатая константа нестойкости(устойчивости).
Ступенчатые константы нестойкости этих комплексов равняются соответственно &. Высокие значения & Г1 и k2 — l свидетельствуют о том, что при образовании комплексов с медью ( II) этилендиамин выступает как полидентатный лиганд. Произведение ступенчатых констант нестойкости равно общей константе нестойкости, индекс которой показывает, из каких множителей она образовалась.Зная величины ступенчатых констант нестойкости ( в частности константы, отвечающей отщеплению первой молекулы аммиака или амина от комплексного иона), концентрацию комплекса, константу RH как основания и константу Me. Зная функцию закомплексованности, ступенчатые константы нестойкости вычисляются методом Ледена. Так, при рассмотрении ступенчатых констант нестойкости комплексных аммиакатов Ni ( II), Cu ( II) пли Со ( П) видно, что для этих солей сольватацпонное равновесие с отщеплением свободного основания будет выражено гораздо сильнее, чем усиление степени кислотной диссоциации аммиака пли аминов в поле двухвалентных ионов. Так, при рассмотрении ступенчатых констант нестойкости комплексных аммиакатов Ni ( II) Cu ( II) или Со ( П) видно, что для этих солей сольватационное равновесие с отщеплением свободного основания будет выражено гораздо сильнее, чем усиление степени кислотной диссоциации аммиака или аминов в поле двухвалентных ионов.]
Эл 1, Рп-называют ступенчатыми константами нестойкости.
Рп ] ( Рп называют ступенчатыми константами нестойкости.
Значение общей константы нестойкости равно произведению значений всех ступенчатых констант нестойкости. [9]
Последние применительно к диссоциации комплексных ионов называются их ступенчатыми константами нестойкости
А — ион водорода или адденд, Ki — кажущаяся ступенчатая константа нестойкости комплексного соединения МА, которая может считаться в первом приближении величиной постоянной только при постоянной ионной силе раствора. Термодинамические уравнения, описывающие соответствующие процессы, идентичны для обоих процессов. [11По кривой комплексообразования, как было показано Бьеррумом, можно определить ступенчатые константы нестойкости аммиакатов меди с различным числом молекул МНз в координационной сфере и, зная их, рассчитать концентрации комплексов различного состава при любых концентрациях свободного аммиака в растворе. [12]Каждой стадии диссоциации комплекса соответствует ступенчатая константа диссоциации, которую называют ступенчатой константой нестойкости и обозначают йнест. Чем сильнее диссоциирует комплекс, тем большее значение имеет / г ест — Константы нестойкости используют для характеристики устойчивости любой комплексной частицы в растворе независимо от того, какие лиганды она отщепляет. [13]Каждой стадии диссоциации комплекса соответствует ступенчатая константа диссоциации, которую называют ступенчатой константой нестойкости и обозначают йвест. Чем сильнее диссоциирует комплекс, тем большее значение имеет Анест. Константы нестойкости используют для характеристики устойчивости любой комплексной частицы в растворе независимо от того, какие лиганды она отщепляет. [14]
Дата добавления: 2015-12-26 ; просмотров: 4503 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Коллигативные свойства растворов
Любому раствору характерны те или иные физические свойства, к которым относятся и коллигативные свойства растворов. Это такие свойства, на которые не оказывает влияние природа растворенного вещества, а зависят они исключительно от количества частиц этого растворенного вещества.
К коллигативным свойствам растворов относятся:
- Понижение давление паров
- Повышение температуры кипения
- Понижение температуры затвердевания (кристаллизации)
- Осмотическое давление раствора.
Рассмотрим подробнее каждое из перечисленных свойств.
Понижение давления паров
Давление насыщенного пара (т.е. пара, который пребывает в состоянии равновесия с жидкостью) над чистым растворителем называется давлением или упругостью насыщенного пара чистого растворителя.
Если в некотором растворителе растворить нелетучее вещество, то равновесное давление паров растворителя при этом понижается, т.к. присутствие какого – либо вещества, растворенного в этом растворителе, затрудняет переход частиц растворителя в паровую фазу.
Экспериментально доказано, что такое понижение давления паров напрямую зависит от количества растворенного вещества. В 1887 г. Ф.М. Рауль описал количественные закономерности коллигативных свойств растворов.
Первый закон Рауля
Первый закон Рауля заключается в следующем:
Давление пара раствора, содержащего нелетучее растворенное вещество, прямо пропорционально мольной доле растворителя в данном растворе:
p — давление пара над раствором, Па;
p0 — давление пара над чистым растворителем, Па;
χр-ль — мольная доля растворителя.
nв-ва и nр-ля – соответственно количество растворенного вещества и растворителя, моль.
Иногда Первому закону Рауля дают другую формулировку:
относительное понижение давления насыщенного пара растворителя над раствором равно мольной доле растворенного вещества:
При этом принимаем, что χв-ва + χр-ль= 1
Изотонический коэффициент Вант-Гоффа
Для растворов электролитов данное уравнение приобретает несколько иной вид, в его состав входит изотонический коэффициент i:
Δp — изменение давления паров раствора по сравнению с чистым растворителем;
i – изотонический коэффициент.
Изотонический коэффициент (или фактор Вант-Гоффа) — это параметр, не имеющий размерности, который характеризует поведение какого – либо вещества в растворе.
То есть, изотонический коэффициент показывает, разницу содержания частиц в растворе электролита по сравнению с раствором неэлектролита такой же концентрации. Он тесно связан связан с процессом диссоциации, точнее, со степенью диссоциации и выражается следующим выражением:
n – количество ионов, на которые диссоциирует вещество.
α – степень диссоциации.
Повышение температуры кипения или понижение температуры затвердевания (кристаллизации). Второй закон Рауля
Равновесное давление паров жидкости имеет тенденцию к увеличению с ростом температуры, жидкость начинает кипеть, при уравнивании давления ее паров и внешнего давления.
При наличии нелетучего вещества, давление паров раствора снижается, и раствор будет закипать при более высокой температуре, по сравнению с температурой кипения чистого растворителя.
Температура замерзания жидкости также определяется той температурой, при которой давления паров жидкой и твердой фаз уравниваются.
Ф.М. Рауль доказал, что повышение температуры кипения, так же как и понижение температуры замерзания разбавленных растворов нелетучих веществ, прямо пропорционально моляльной концентрации раствора и не зависит от природы растворённого вещества. Это правило известно как Второй закон Рауля:
K — криоскопическая константа,
mв-ва — моляльность вещества в растворе.
Растворы электролитов не подчиняются Законам Рауля. Но для учёта всех несоответствий Вант-Гофф предложил ввести в приведённые уравнения поправку в виде изотонического коэффициента i, учитывающего процесс распада на ионы молекул растворённого вещества:
Осмотическое давление раствора
Некоторые материалы имеют способность к полупроницаемости, т.е. им свойственно пропускать частицы определенного вида и не пропускать частицы другого вида.
Перемещение молекул растворителя (но не растворенного, в нем вещества), через полупроницаемую мембрану в раствор с большей концентрацией из более разбавленного представляет собой такое явление как осмос.
Представим два таких раствора, которые разделены полупроницаемой мембраной, как показано на рисунке выше. Растворы стремятся к выравниванию концентраций, поэтому вода будет проникать в раствор, тем самым уменьшая его концентрацию.
Для того, чтобы осмос приостановить, необходимо приложить внешнее давление к раствору. Такое давление, которое требуется приложить, называется осмотическим давлением.
Осмотическое давление и концентрацию раствора неэлектролита позволяет связать уравнение Вант — Гоффа, которое напоминает уравнение идеального газа Клапейрона – Менделеева:
где C — молярная концентрация раствора, моль/м 3 ,
R — универсальная газовая постоянная (8,314 Дж/моль·К);
T — абсолютная температура раствора.
Преобразуем уравнение следующим образом:
C = n/V = m/(M·V)
π = т·R·T / M·V или
Для растворов электролитов осмотическое давление определяется уравнением, в которое входит изотонический коэффициент:
где i — изотонический коэффициент раствора.
Для растворов электролитов i > 1, а для растворов неэлектролитов i = 1.
Если полупроницаемой перегородкой разделены два раствора, имеющие одинаковое осмотическое давление, то перемещение растворителя через перегородку отсутствует. Такие растворы называются изотоническими.
Раствор, с меньшим осмотическим давлением, по сравнению с более концентрированным раствором, называют гипотоническим, а раствор с большей концентрацией – гипертоническим.
ВАНТ-ГОФФА ПРАВИЛО
ВАНТ-ГОФФА ПРАВИЛО. Почти все химические реакции при повышении температуры идут быстрее. Зависимость скорости реакции от температуры описывается уравнением Аррениуса:
k = Ae –E a/RT, где k – константа скорости реакции, А – не зависящая от температуры константа (ее называют предэкспоненциальным множителем), Еа– энергия активации, R – газовая постоянная, Т – абсолютная температура. В школьных учебниках зависимость скорости реакции от температуры определяют в соответствии с так называемым «правилом Вант-Гоффа», которое в 19 в. сформулировал голландский химик Якоб Вант-Гофф. Это чисто эмпирическое правило, т.е. правило, основанное не на теории, а выведенное из опытных данных. В соответствии с этим правилом, повышение температуры на 10° приводит к увеличению скорости в 2–4 раза. Математически эту зависимость можно выразить уравнением v2v1 = g (T2 – T 1 )/10 , где v1 и v2– скорости реакции при температурах Т1 и Т2; величина g называется температурным коэффициентом реакции. Например, если g = 2, то при Т2– Т1 = 50 о v2/v1 = 2 5 = 32, т.е. реакция ускорилась в 32 раза, причем это ускорение никак не зависит от абсолютных величин Т1 и Т2, а только от их разности.
Однако из уравнения Аррениуса следует, что температурный коэффициент реакции зависит как от энергии активации, так и от абсолютной температуры. Для данной реакции с определенным значением Еа ускорение при повышении температуры на 10° будет тем больше, чем ниже температура. Это почти очевидно и без расчетов: повышение температуры от 0 до 10° С должно сказаться на скорости реакции значительно сильнее, чем такое же повышение температуры, например, от 500 до 510° С.
С другой стороны, для данного температурного интервала ускорение реакции будет тем сильнее, чем больше ее энергия активации. Так, если энергия активации реакции мала, то такая реакция идет очень быстро, и при повышении температуры на 10° С ее скорость почти не изменяется. Для таких реакций температурный коэффициент намного меньше 2. Для реакций же с большой энергией активации, которые при невысоких температурах идут медленно, ускорение при повышении температуры на 10° С может значительно превысить 4-кратное.
Например, реакция диоксида углерода со щелочным раствором с образованием гидрокарбонат-иона (СО2 + ОН® НСО3 – ) имеет энергию активации 38,2 кДж/моль, поэтому при повышении температуры, например, от 50 до 60° С эта реакция ускорится всего в 1,5 раза. В то же время реакция распада этилбромида на этилен и бромоводород (С2Н5Вr ® С2Н4 + НВr) с энергией активации 218 кДж/моль ускорится при повышении температуры от 100 до 110 o С в 6,3 раза (правда, в этом интервале температур реакция идет очень медленно). Кинетика реакции атомов водорода с этаном H + C2H6 ® H2 + C2H5 была изучены в широком температурном интервале – от 300 до 1100 К (27–827° С). Для этой реакции Eа = 40,6 кДж/моль. Следовательно, повышение температуры на 10° вызовет увеличение скорости реакции в 1,7 раза в интервале 300–310 K и только в 1,04 раза в интервале 1090–1100 K. Так что при высоких температурах скорость этой реакции практически не зависит от температуры. А для реакции присоединения атома водорода к двойной связи H + C2H4 ® C2H5 энергия активации мала (Eа = 3,4 кДж/моль, так что ее скорость слабо зависит от температуры в широком температурном интервале. И только при температурах намного ниже 0° С начинает сказываться наличие активационного барьера.
Подобных примеров можно привести множество. Очевидно, что правило Вант-Гоффа противоречит не только уравнению Аррениуса, но и многим экспериментальным данным. Откуда же оно взялось и почему нередко выполняется?
Если в приведенном выше математическом выражении для правила Вант-Гоффа подставить вместо скоростей v1 и v2 для данной реакции их зависимости от температуры, в соответствии с уравнением Аррениуса, то после сокращения предэкспоненциальных множителей получим следующее выражение: g = vT +10/vT = е –Е а/R(Т+10)/е –Е а/RТ = е (Еа/R)[1/Т – 1/(T+10)] . Логарифмироване этого уравнения дает: lng = (Eа/R)[1/T – 1/(T + 10)], откуда Еа = Rlng T(T + 10)/10 = 0,83lngT(T + 10). Энергия активации не является функцией температуры, эта зависимость нужна лишь для удобства последующего анализа. Последнее уравнение – это уравнение параболы, в котором физический смысл имеют только положительные значения. Соответствующая диаграмма ограничена двумя ветвями параболы: при g = 2 получаем Еа = 0,58Т(Т + 10), при g = 4 получаем Еа = 1,16Т(Т + 10). К тем же формулам приходим и при использовании десятичных логарифмов. Соответствующие графики двух парабол, для значений g 2 и 4, приведены на рисунке. Их физический смысл заключается в том, что области выполнения правила Вант-Гоффа соответствует только область между параболами. Таким образом, существуют только определенные соотношения между энергией активации реакции и температурой ее проведения, при которых правило Вант-Гоффа выполняется. Ниже нижней ветви температурный коэффициент g 4.
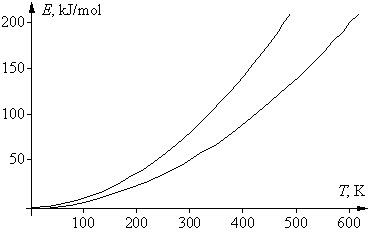
Если посмотреть, какие реакции «укладываются» в указанную довольно узкую область, то окажется, что все эти реакции идут не слишком быстро и не слишком медленно, а с удобной для измерения (при данной температуре) скоростью. Скорость только таких реакций и могли изучать химики во времена Вант-Гоффа. Например, если энергия активации была невелика (меньше 50 кДж/моль), то такая реакция при комнатной температуре заканчивалась за 1–2 секунды; поэтому для изучения ее кинетики следовало значительно понизить температуру, чтобы реакция проходила не быстрее, чем за 10–20 минут. Только в этом случае химики 19 в. успевали отбирать пробы по ходу реакции и анализировать изменение в них концентрации реагентов. Других способов изучения скорости реакции в то время не было. Если это не удавалось (например, водный раствор замерзал), то скорость такой реакции не изучали. Если же энергия активации реакции была велика и при комнатной температуре она шла слишком медленно (многие сутки, или даже недели), то температуру повышали, чтобы реакция шла с удобной для измерения скоростью. И здесь были свои ограничения – например, раствор мог закипеть, т.е. в любом случае исследователи фактически «подстраивали» изучаемую реакцию под область между двумя параболами.
Сейчас химики имеют возможность с помощью различных приборов экспериментально изучать и очень быстрые (идущие в микросекундной области), и очень медленные реакции, для которых температурный коэффициент может быть значительно меньше 2 или значительно больше 4. Поэтому правило Вант-Гоффа, которое, в отличие от уравнения Аррениуса, не имеет четкого физического смысла, представляет лишь чисто исторический интерес и в современной науке не используется. В подавляющем большинстве учебников и монографий по химической кинетике, а также в 5-томной Химической Энциклопедии это правило даже не упоминается. И, тем не менее, если изучаемая реакция идет с удобной для измерения скоростью, например, заканчивается за 30–40 мин, а энергия активации ее еще не измерена, то для предварительной грубой оценки зависимости скорости такой реакции от температуры можно использовать правило Вант-Гоффа. Поэтому это правило приводится во всех школьных учебниках химии.
http://zadachi-po-khimii.ru/obshaya-himiya/kolligativnye-svojstva-rastvorov.html
http://www.krugosvet.ru/enc/nauka_i_tehnika/himiya/VANT-GOFFA_PRAVILO.html