Химия : Квантово-химические правила отбора элементарных стадий
Квантово-химические правила отбора элементарных стадий
Любая термодинамически разрешенная реакция, в которой происходит незначительное перемещение ядер (близость минимумов энергетических термов) и мало изменяются электронные состояния (принцип наименьшего движения), и молекулярность которой не превышает 2, имеет шанс быть согласованным процессом, элементарной стадией. Однако, для того, чтобы величина была небольшой и реакция протекала с измеряемой скоростью, необходимо выполнение двух требований, вытекающих из квантово-химической теории. Эффективное взаимодействие двух частиц с достаточно низкой величиной барьера может происходить в случае, когда симметрия перекрывающихся молекулярных орбиталей (МО) будет одинаковой, а энергии этих МО будут близки. Например, бимолекулярная реакция (27)
с небольшим изменением координат ядер и валентных оболочек не является элементарным процессом (ЭС), поскольку запрещена по симметрии граничных МО. Рассмотрим подробнее некоторые квантово-химические подходы к проблеме реакционной способности.
Теория возмущений в приближении граничных МО
Из правила БЭП следует, что знание энергетического состояния исходных и конечных продуктов позволяет оценивать кинетические характеристики ЭС (вероятность реализации элементарного акта). Метод возмущения МО (МВМО), оперируя только граничными занятыми и свободными МО (ВЗМО, НСМО) и зарядами (на атомах в молекулах и на атомных орбиталях в МО) в исходных реагентах, позволяет в ряде случаев предсказать вероятность, направление и эффективность взаимодействия двух реагентов.
Чем эффективнее взаимодействие, тем ниже Еакт и тем выше вероятность согласованного (элементарного) акта.
Если энергии граничных орбиталей 1 и 2 близки, то энергия взаимодействия определяется резонансным (обменным) интегралом 12
где H — гамильтониан системы, — элемент объема, в котором происходит перекрывание орбиталей. Величина 12 в этом случае определяет и величину расщепления новых МО 1 и 2 или энергию стабилизации = 12.
Если энергии 1 и 2 различаются сильно, то величина определяется не только 12, а зависит и от разности энергий 1 и 2 по уравнению (29):
где Е1 и Е2 — энергии низшей и высшей МО
Чем больше величина , тем стабильнее образующийся аддукт, тем ниже Еакт его образования.
МВМО не дает оценки Е переходного состояния и Еакт. Рассчитывается лишь разница между полной электронной энергией реагирующей системы Е и энергиями исходных реагентов и (малое возмущение):
справедливое только для начальных участков координаты реакции. Только на больших расстояниях между реагентами не происходит смешения МО, нет межмолекулярного отталкивания и можно говорить о чистых МО исходных реагентов. Вместе с тем, такое приближение позволяет оценить наиболее вероятный путь реакции.
Энергию возмущения Е при взаимодействии реагентов S и Т (S и Т — молекулы или активные центры в молекулах) рассчитывают по уравнению (31):
В случае только двух граничных МО (например, молекул донора и акцептора) уравнение упрощается (32):
В уравнениях (31, 32) qS и qT — эффективные заряды на центрах S и Т, RST — расстояние между центрами в ходе взаимодействия, — диэлектрическая проницаемость среды. Таким образом, первый член (возмущение 1го порядка) отражает энергию кулоновского взаимодействия. Второй член (возмущение 2го порядка) определяет энергию орбитального перекрывания и включает: ST — коэффициент, учитывающий заселенность электронами орбиталей 1 и 2, и — квадраты коэффициентов при атомных орбиталях центров S и Т волновой функции граничных МО 1 и 2, — квадрат обменного интеграла, Е1 и Е2 — энергии орбиталей 1 и 2. Разные случаи заселенности орбиталей 1 и 2 реагирующих частиц и коэффициент ST приведены ниже:
Число электронов на
граничных орбиталях
0 (нет перекрывания)
2 (самое сильное перекрывание)
Если Е1 — Е2 в знаменателе уравнения (32) мало, заселенность ST равна 1 и 2, симметрия орбиталей одинакова ( 12 > 0), геометрия орбиталей удобна для перекрывания (коэффициенты CS и CT имеют большие значения в одинаковых областях пространства) и второй член существенно больше первого, можно говорить об орбитально-контролируемой реакции.
Если Е1 — Е2 величина большая, второй член становится небольшим даже при больших CS и CT. Если при этом qS и qT также велики, говорят о зарядово-контролируемой реакции. Эти простые оценки полуэмпирическими методами МО ЛКАО позволяют определить (без расчетов ППЭ), в каком направлении (по каким центрам) пойдет та или другая реакция и можно ли ожидать высокой скорости от выбранной элементарной стадии. Естественно, что все соображения об оценке энергии Е относятся только к элементарным стадиям.
Предположим, что донорная молекула, типичный нуклеофил SCN- реагирует с акцептором, имеющим НСМО. Если энергии ВЗМО донора ( 1) и НСМО акцептора ( 2) близки, реакция будет орбитально-контролируемой. Такая реакция будет протекать между молекулой акцептора и тем центром нуклеофила (донора), который обладает наивысшей плотностью заряда () на граничных орбиталях донора. Высшая занятая МО нуклеофила SCN- 2 имеет вид:
2 = 0.74 S + 0.33 C — 0.59 N
Поэтому реакция с акцептором пойдет через атом S
Если орбиталь акцептора лежит высоко и Е1 — Е2 велика, реакция контролируется зарядовым взаимодействием. В этом случае, первый (кулоновский) член в уравнении (32) будет больше для того центра нуклеофила, у которого выше qi. Для расчета qS и qN необходимо учесть коэффициент при этих центрах на всех орбиталях, т.е. кроме 2 нужно учесть и НЗМО 1
1 = 0.33 S + 0.59 C + 0.74 N
qi рассчитывается по уравнению
qS = 1 — 2(0.332 + 0.742) = — 0.313
qN = 1 — 2(0.592 + 0.742) = — 0.7914
т.е. в анионе на атоме N эффективный отрицательный заряд выше |qN| > |qS| (заряд на атоме С, qС 0.1). Таким образом, в условиях кулоновского контроля нуклеофил SCN- будет взаимодействовать с акцептором атомом азота
МВМО дал теоретическое объяснение ряду эмпирических правил и обобщений. В 1958 г Арланд, Чатт и Дэвис предложили классификацию комплексов металлов, разделив их на две группы (а) и (б). К группе (а) были отнесены ионы металлов (в наиболее распространенных степенях окисления), которые образуют наиболее устойчивые комплексы с лигандами, имеющими донорные атомы N, O, F. К группе (б) они отнесли ионы, образующие наиболее стабильные комплексы с лигандами, содержащими донорные атомы элементов третьего и последующих периодов (P, S, Cl, Br, J). Так, например, устойчивость галогенидных комплексов Zn2+ (группа (а)) и Hg2+ (группа (б)) меняется в следующих рядах:
Zn2+ F- >> Cl- > Br- > I-
Hg2+ I- > Br- > Cl- >> F-
При переходе к Hg2+ происходит обращение ряда устойчивости по сравнению с “обычным” рядом (Zn2+), согласующимся с простыми электростатическими представлениями.
Очевидно, что в случае первой группы ионов определяющим является зарядовый, а в случае второй группы ионов — орбитальный фактор. Аналогичные объяснения получили правило взаимодействия жестких и мягких кислот и оснований (Пирсон, 1963) и правило Корнблюма.
В терминах теории Пирсона взаимодействие жестких частиц (кислот и оснований, акцепторов и доноров) соответствует зарядовому контролю, взаимодействие мягких частиц — орбитальному контролю. Степень жесткости и мягкости акцептора (A) и донора (D) можно оценивать по различным критериям. Приведем величины орбитальных электроотрицательностей En(A) Em(D) (в эВ) по Клопману:
В приведенной таблице самая жесткая кислота — Al3+, самое жесткое основание — F-. Самая мягкая кислота — Hg2+, самое мягкое основание — H-.
Орбитальная симметрия и правила отбора
Общие правила отбора ЭС по симметрии МО в реагирующей системе с циклическим многоцентровым переходным состоянием сформулировали Р.Вудворд и Р.Хоффман — правила сохранения орбитальной симметрии в ходе согласованных реакций.
Если заполненные связывающие МО реагентов коррелируют по симметрии (имеют одинаковую симметрию) с заполненными связывающими МО продуктов реакции, такая реакция будет идти согласованно термически (как ЭС). В ходе такой реакции симметрия взаимодействующих орбиталей сохраняется вдоль координаты реакции по ППЭ. Если такой корреляции нет, согласованная реакция пойдет только фотохимически.
В простых молекулах анализ симметрии граничных орбиталей позволяет сделать заключение о возможности согласованной ЭС. Например, симметрии занятой -МО молекулы Н2 и свободной *-МО молекулы I2 не позволяют реализоваться циклическому переходному состоянию
Это же касается и разрыхляющей *-МО H2 и высшей занятой -МО I2. Граничные ВЗМО и НСМО двух молекул этилена имеют разную симметрию и не могут образовать 4-членного переходного состояния при протекании ЭС
Занятая -МО одной молекулы этилена
не может перекрываться синхронно со свободной *-МО второй молекулы. Симметрия этих МО различна (относительно плоскости, проходящей перпендикулярно связи С-С через ее центр). В реакции бутадиена с этиленом, НСМО C4H6 (1*-C4H6) имеет одинаковую симметрию с ВЗМО C2H4 и процесс протекает по согласованному 6-центровому механизму
Аналогично и для перекрывания *-C2H4 и НЗМО C4H6 (2-C4H6).
Запрещенными по симметрии как элементарные стадии являются реакции присоединения молекул H2, Cl2, HCl, HF, HCN к кратным связям олефинов и алкинов (через 4-членное циклическое переходное состояние).
Реакции нуклеофильного и электрофильного присоединения и замещения, протекающие через линейные переходные состояния разрешены по симметрии. Участие переходных металлов (d-орбитали и d-электроны) в ЭС снимает запреты по симметрии и делает реакции согласованного присоединения по кратным связям металлосодержащих фрагментов разрешенными ЭС.
Разрешены по симметрии орбиталей также реакции присоединения молекул НХ к координированным атомом металла алкенам.
Правило сохранения 16-18 электронной оболочки Толмена в элементарных стадиях
Уже давно было отмечено (Сиджвик, 1929), что в стабильных комплексных соединениях общее количество электронов вокруг атома металла равно числу электронов ближайшего инертного газа. Это число электронов было названо эффективным атомным номером (ЭАН). В случае d-металлов число электронов в валентной оболочке металла, связанного с лигандами, должно быть равно 18 (d10s2p6). Такая оболочка и считается устойчивой. Например, Ni(CO)4: Ni0 d10, CO — 2-х электронный лиганд. Следовательно, 10 + 8 = 18. Для расчета числа электронов в комплексе металла необходимо сложить число электронов в валентной оболочке атома металла (или иона) и число электронов, предоставляемых нейтральными лигандами (или анионами). Для этого используют ковалентную и ионную модели химической связи. В первом случае комплекс включает ионы Mn+, X- и нейтральные лиганды L, а во втором — атомы металла, нейтральные группы X (гомолитический разрыв связи M-X) и нейтральные лиганды L. Например, в комплексе HMn(CO)5 в валентной оболочке Mn имеем для ионной модели:
H- (2 эл) + Mn+ (6 эл) + 5CO (10 эл) = 18 эл.
для ковалентной модели:
H· (1 эл) + Mn0 (7 эл) + 5CO (10 эл) = 18 эл.
В таблице 2.1 приведены некоторые лиганды, их обозначения и количества электронов, предоставляемых металлу в рамках ковалентной и ионной моделей.
1.3. РАСЧЕТ ИНДЕКСОВ РЕАКЦИОННОЙ СПОСОБНОСТИ
Для некоторых реакций наблюдается хорошая корреляция между выходами конечных продуктов и так называемыми индексами реакционной способности. В качестве индексов обычно используют заряды на атомах, порядки связей, энергии граничных МО, квадраты коэффициентов разложения граничных МО по базису АО (парциальные электронные плотности) и т.д. Здесь термином «граничные МО» обозначены верхняя занятая и нижняя вакантная МО соединения. Наиболее часто пользуются зарядами на атомах и параметрами граничных МО, поэтому мы остановимся только на этих индексах реакционной способности.
Почему же имеют место корреляции между индексами реакционной способности и выходом конечных продуктов реакций? Вообще говоря, выходы конечных продуктов реакции определяются свободными энергиями активации, а индексы реакционной способности характеризуют энергию межмолекулярных взаимодействий для достаточно удаленных друг от друга реагентов. Однако для некоторых реакций эти параметры (свободные энергии активации и энергии межмолекулярных взаимодействий) количественно связаны друг с другом, т.e. с увеличением энергии взаимодействия между реагентами свободная энергия активации уменьшается.
Энергию межмолекулярного взаимодействия при сближении реагентов можно условно разбить на вклады трех типов: кулоновские, орбитальные и стерические. Энергия кулоновского взаимодействия зависит от распределения электронной плотности или от зарядов на атомах реагентов. Поэтому для некоторых реакций удается найти корреляцию между зарядами на атомах и выходом конечных продуктов реакции. Так, нуклеофильные реагенты (атакующий центр заряжен отрицательно) присоединяются преимущественно к атомам, на которых локализованы большие положительные заряды, а электрофильные (атакующий центр заряжен положительно), наоборот, — к атомам, на которых локализованы большие отрицательные заряды.
Корреляции между выходом конечных продуктов реакции и зарядами на атомах широко используются химиками-синтетиками для объяснения экспериментальных данных, и здесь все более или менее ясно. Отметим только, что понятие заряда на атоме имеет условный характер. На атоме в действительности локализован лишь заряд ядра, электроны внутренних оболочек находятся вблизи ядер, а валентные электроны локализованы между атомами. Обычно при вычислении заряда на атоме в квантовой химии пользуются анализом электронных заселенностей, предложенным Малликеном [31]. В этом приближении заряд qA на атоме А вычисляется по следующей формуле:
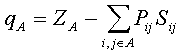
Здесь сумма берется по всем орбиталям i и j атома A; ZA — заряд ядра;
Рij— матрица зарядов и порядков связей; Sij— матрица интегралов перекрывания. В полуэмпирических методах обычно пользуются упрощенной формулой 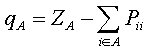
Величины зарядов на атомах, вычисленные в этом приближении, в неэмпирических расчетах очень сильно зависят от выбора базиса, а в полуэмпирических — от выбора метода. Заряды на атомах, вычисленные в разных базисах (неэмпирические расчеты) и разными методами (полуэмпирические расчеты), могут различаться в 1,5—2 раза, но качественные результаты (знак и относительная величина заряда) обычно остаются одинаковыми. В неэмпирических расчетах заряды на атомах при расширении базиса обычно увеличиваются по абсолютной величине.
Формально заряды на атомах в приближении Малликена можно вычислить в любом базисе, но при включении в него сильно диффузных орбиталей результаты становятся сомнительными, так как атому приписывается электронная плотность, которая в действительности от него сильно удалена. В частности, не следует использовать анализ заселенностей по Малликену при включении в базис поляризационных орбиталей (расчеты в базисах 6-31ГФ* и 6-31ГФ**) и диффузных s- и p-орбиталей (расчеты в базисе 3-21+ГФ). Отметим также, что заряды на атомах, вычисленные методом ППДП/2, обычно близки к результатам неэмпирического расчета в минимальном базисе, а методы МЧПДП/3 и МПДП дают приблизительно такое же распределение электронной плотности, как неэмпирические расчеты в валентно-расщепленных базисах.
При использовании зарядов на атомах для изучения реакций, которые идут в одну стадию или у которых первая стадия определяет направление и выход конечных продуктов, достаточно рассчитать электронную структуру исходных реагентов и провести корреляцию между вычисленными зарядами на атомах и направлением реакции. Такие работы хорошо известны. В частности, так обычно объясняют влияние заместителей на направление реакций нуклеофильного и электрофильного замещения ароматических соединений. Однако для большинства реакций подобные корреляции провести не удается. Иногда можно найти корреляцию между электронным строением интермедиата, который образуется на одной из элементарных стадий, и выходами конечных продуктов реакции. В этом случае квантово-химические расчеты приходится проводить для различных метастабильных промежуточных продуктов и отбирать из них интермедиаты, электронная структура которых позволяет объяснить экспериментально наблюдаемое направление реакции. Так, на основе расчета электронной структуры тиофеновых α,β-непредельных кетонов в работах [32, 33] не удалось объяснить направление реакции нитрования в сильно кислых средах, поэтому пришлось провести расчеты для протонированных форм, установить, какие интермедиаты, образующиеся при протонировании, являются наиболее устойчивыми, и лишь на основе анализа их электронной структуры удалось найти корреляцию между направлением нитрования и зарядами на атомах. Однако для большинства реакций подобные корреляции найти не удается. Под термином «орбитальное взаимодействие» понимают взаимодействие между молекулярными орбиталями реагентов при их сближении. Это взаимодействие имеет квантово-механическое происхождение. По своей природе оно близко к хорошо известному химикам эффекту сопряжения. Рассмотрим механизм орбитального взаимодействия на качественном уровне. При сближении реагентов Х и Y между их орбиталями φX и φYвозникает перекрывание и, как следует из квантовой теории, разность энергий E(φX)-E(φY) увеличивается по абсолютной величине.
Пусть E(φX)>, тогда взаимодействие между φ X и φYприведет к уменьшению E(φX) и увеличению E(φY) на одинаковые значения (рис. 1.2). Если φX и φY— занятые орбитали соединений Х и Y, то полная энергия системы X. Y не изменится (рис. 1.2,а), но если φX— занятая орбиталь, а φY— вакантная, то произойдет понижение полной энергии реагентов (рис. 1.2,б), другими словами, между реагентами появится орбитальное взаимодействие. Таким образом, при сближении реагентов Х и Y взаимодействие занятых МО φXi и вакантных МО φ*Ykприводит к понижению положения занятых МО φXi на шкале энергий и к стабилизации системы X. Y. К аналогичному результату приводит взаимодействие занятых МО φYkи вакантных МО φ*Xi.
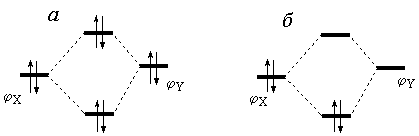
Рис. 1.2. Орбитальное взаимодействие реагентов Х и У
Энергию орбитального взаимодействия можно оценить во втором порядке теории возмущений [2]:
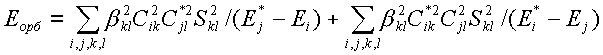
Здесь сумма по i берется по всем занятым (первое слагаемое) и всем вакантным (второе слагаемое) МО X, сумма по jберется по всем вакантным (первое слагаемое) и занятым (второе слагаемое) МО Y, сумма по k берется по всем АО X, по l — по всем АО Y; βkl— коэффициент, имеющий размерность энергии; Сik и Сjl— коэффициенты разложения МО Х и Y по базисным АО; Sij — интеграл перекрывания; Е*i, E*j, Еi и Ej — энергии МО φ*Xi, φ*Yj, φXiи φYj.
Стабилизация системы X. Y за счет орбитального взаимодействия любых пар МО обратно пропорциональна разности их энергий, т.е. чем дальше друг от друга лежат орбитали на шкале энергий, тем слабее они взаимодействуют. Поэтому на практике обычно пользуются приближением граничных орбиталей, т.е. учитывают взаимодействие лишь между двумя МО, для которых разность энергий минимальна. В этом приближении энергия орбитального взаимодействия зависит от энергий граничных МО и от коэффициентов разложения этих МО по базису АО. Любую из этих величин можно использовать в качестве индекса реакционной способности, но наиболее часто пользуются разностью энергий граничных МО. Так, в работах [34, 35] при изучении реакции Дильса—Альдера циклических диенов (циклопентадиена, гексахлорциклопентадиена и тетрахлор-1,2-бензохинона) с диенофилами (монозамещенными ацетиленами, сопряженными аминами, сопряженными диенами и триенами) была обнаружена корреляция между выходами конечных продуктов реакции и положением граничных МО на шкале энергий. Этот результат свидетельствует, что для данной реакции определяющую роль играет орбитальное взаимодействие.
Индексы реакционной способности весьма широко применяются в прикладной квантовой химии, однако с их помощью можно решать лишь весьма ограниченный круг вопросов. В большинстве случаев они не позволяют определить ни направление, ни относительную скорость реакции, поэтому для изучения реакционной способности органических соединений приходится применять более сложные методики (расчеты тепловых эффектов и поверхностей потенциальной энергии). В качестве примера рассмотрим результаты работы [36], в которой был изучен механизм присоединения СН-кислот типа XCH2CО2Et (X = CO2Et, COMe, CN) к α,β-непредельным альдегидам в условиях межфазного катализа. Из эксперимента было известно, что эта реакция может идти по двум направлениям: карбанион, генерированный из СН-кислоты, может присоединяться к карбонильному атому углерода и к β-атому углерода связи С=С.
Предполагалось, что в первом случае направление реакции определяется электростатическим взаимодействием (зарядовый контроль), а в во втором — орбитальным (орбитальный контроль). Влияние заместителей на направление присоединения при этом объясняли изменением относительной величины электростатического и орбитального взаимодействий. Однако результаты квантово-химических расчетов [36] показали, что это не так. Оказалось, что такие заместители, как хлор, метильная и фенильная группы, практически не меняют относительную величину орбитального и электростатического взаимодействий, хотя и меняют направление присоединения.
Все попытки объяснить влияние заместителей на направление реакции с помощью статических индексов реакционной способности окончились безрезультатно. Дальнейшее исследование показало, что в данном случае на направление присоединения основное влияние оказывает стерический эффект. Мы специально привели этот пример, так как при изучении присоединения заряженного реагента к ненасыщенному атому углерода, как правило, удается найти корреляцию между направлением присоединения и индексами реакционной способности (обычно π- или π+σ-электронными зарядами на атомах), но даже и здесь, как видим, есть исключения. Для других классов органических реакций область применения индексов реакционной способности уменьшается.
В заключение этого раздела подчеркнем еще одно очень важное обстоятельство. При поиске корреляций между индексами реакционной способности и выходами продуктов реакции необходимо располагать достаточно большим материалом для сравнения. Если имеются данные лишь для 5—7 родственных соединений, то статистическая вероятность сделать ошибочное заключение будет очень велика. Кроме того, при поиске корреляций между результатами квантовохимических расчетов для газофазных моделей и данными эксперимента, полученными в растворе, необходимо помнить, что растворитель очень сильно меняет электронную структуру ионов, при этом наиболее значительно меняются энергии МО.
Теоретическое обоснование принципа жестких и мягких кислот и оснований
Вы будете перенаправлены на Автор24
Количественное описание принципа ЖМКО
Для количественного описания жестких и мягких кислот и оснований используют значения абсолютной электроотрицательности и абсолютной жесткости молекул.
Абсолютную электроотрицательность молекулы ($\chi $) можно охарактеризовать по положению средней точки между уровнями нижней свободной молекулярной орбитали ($HCMO$) и верхней занятой молекулярной орбитали (ВЗМО).
Абсолютная жесткость ($G$) — это величина, характеризующая энергетические щели между граничными молекулярными орбиталями молекул.
Чем выше абсолютная электроотрицательность, тем электроотрицательней молекула; чем выше значение абсолютной жесткости, тем «жестче» молекула.
При взаимодействии кислоты с основанием осуществляется формирование донорно — акцепторного комплекса. Сила кислотно — основного взаимодействия (прочность комплекса) определяется степенью переноса заряда от более электроотрицательной молекулы к менее электроотрицательной.
Степень переноса заряда $\triangle N$ имеет размерность долей электрона и связана с абсолютной электроотрицательностью и абсолютной жесткостью молекулы выражением:
где $A$ — кислота, $B$ — основание, $A$ и $B$ имеют одинаковую жесткость.
Чем больше разность электроотрицательностей между молекулами кислоты и основания, тем больше величина $\Delta N$. Чем больше жесткость взаимодействующих молекул кислоты и основания, тем меньше степень переноса заряда.
Разность электроотрицательностей является термодинамической движущей силой переноса заряда от основания к кислоте. Величину сопротивления переносу заряда определяет сумма абсолютных жесткостей молекул $A$ и $B$, т.е. связана с их поляризуемостью.
Образование аддукта между кислотой и основанием зависит от факторов:
- орбитального взаимодействия (взаимодействия граничных орбиталей);
- электростатического взаимодействия, которое значительно усиливается, если реагенты имеют противоположные заряды;
- пространственных препятствий;
- сольватации (если реакция протекает в растворе).
Готовые работы на аналогичную тему
Взаимодействие граничных орбиталей
В реакции между основанием (донор электронов) и кислотой (акцептор электронов) протекают парные взаимодействия между всеми орбиталями кислоты и основания.
Главный вклад в общую энергию возмущения вносит связь между граничными орбиталями основания и кислоты: высшей занятой молекулярной орбиталью (ВЗМО) донора и нижней свободной молекулярной орбиталью ($HCMO$) акцептора. При качественном рассмотрении кислотно — основного взаимодействия достаточно изучить взаимные возмущения ВЗМО донора и $HCMO$ акцептора, особенно если энергия электростатического взаимодействия, при сравнении двух пар, для каждой пары кислота — основание будет одинакова.
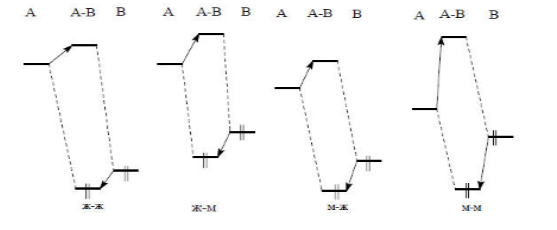
Рисунок 1. Возможные случаи взаимодействия граничных орбиталей при кислотно-основной нейтрализации ($HCMO$ кислоты $A$, ВЗМО основания $B$)
На рис.1 представлены возможные варианты взаимодействия граничных орбиталей в зависимости от взаимного расположения по энергии $HCMO$ кислоты и ВЗМО основания.
Взаимодействия граничных орбиталей не зависят от общего заряда, поэтому их показатели важны как при рассмотрении заряженных, так и незаряженных оснований.
Варианты взаимодействия граничных орбиталей:
Наибольшая стабилизация комплекса $AB$, при взаимодействии граничных орбиталей, следует для основания с относительно высокой энергией ВЗМО и для кислоты с относительно низкой энергией $HCMO$ (взаимодействие «мягкий-мягкий»).
Значения энергии $HCMO$ и ВЗМО близки, т.е. энергия возмущения имеет большую величину. К кислотам с низкой энергией $HCMO$ относятся ионы тяжелых металлов с пустой s-орбиталью ($Ag^+$, $Cu^+$, $Hg^<2+>$) или нейтральные молекулы ($I_2$, $Br_2$, тетрацианоэтилен $(CN)_2C=C(CN)_2)$, а к основаниям с высокой энергией ВЗМО относятся: $CN^-$, $RS^-$, $R_3P$, $CO$ и т.д.
При взаимодействии кислот и оснований по типу «жесткий — жесткий», энергетическое различие между ВЗМО и $HCMO$ очень большое. Поэтому возмущение при взаимодействии этих орбиталей будет совсем слабым. Достаточную стабилизацию комплекса АВ орбитальные связи обеспечить не могут. Данный комплекс может удерживаться только за счет сил электростатического притяжения.
Электростатические взаимодействия
При взаимодействии по типу «мягкий — мягкий» происходит перенос заряда с ВЗМО основания на $HCMO$ кислоты. При взаимодействии между основанием и кислотой проявляется орбитальный контроль. Аддукт стабилизирует кулоновское взаимодействие за счет электростатических сил дальнего действия. В реакции проявляется зарядовый контроль при условии стабильности аддукта (из-за кулоновского взаимодействия).
Когда абсолютные жесткости молекул, вступающих во взаимодействие достаточно большие, проявляется зарядовый контроль для связи по типу «жесткий — жесткий» (Рис. 1).
Согласно теории ЖМКО, жесткие кислоты реагируют с жесткими основаниями, а мягкие кислоты — с мягкими основаниями. Это можно объяснить, исходя из степени взаимодействия в молекуле ВЗМО и $HCMO$ кислоты или основания:
жесткое основание имеет электронную пару, трудно поляризуемую, на низколежащей ВЗМО;
плохая поляризуемость ведет к тому, что электронная пара создает сильное электростатическое поле и прочно удерживается на ВЗМО;
таким же образом, в кислоте, при условии, что первая пустая орбиталь находится высоко, электроны на заполненных орбиталях плохо поляризуемы, в результате чего плотность пустой орбитали большая;
жесткая кислота и жесткое основание характеризуются сильным электростатическим полем и низкой поляризуемостью;
при тесном взаимодействии обоих жестких реагентов происходит сильная стабилизация;
при вовлечении в процесс мягкого реагента, диффузная природа его электронной оболочки приводит к уменьшению электростатического притяжения к противоиону.
Сольватация
Сольватация — это кулоновское взаимодействие между ионами, молекулами растворенного вещества и растворителя.
Малые ионы обладают жесткостью благодаря сольватации протонными растворителями. Протонные растворители имеют плотную «положительную» сольватную оболочку (атомы водорода воды ориентированы к иону), которая способна понижать ВЗМО малых анионов. Повышать энергию НСМО малых катионов будет «отрицательная» сольватная оболочка (атомы кислорода молекулы воды ориентированы к иону).
В результате, жесткость малых ионов будет «приобретенной». Частично малые ионы получают жесткость в начале взаимодействия кислоты и основания при прямом взаимодействии с партнером. В данном случае электростатическое поле данного партнера будет оказывать стабилизирующее влияние, аналогично влиянию протонных растворителей.
Например: Катион лития $Li^+$ в водном растворе можно сделать мягкой кислотой, если ввести его во внутреннюю полость молекулы краун-эфира или криптанда. Имея низкую $HCMO$, ион $Li^+$ будет вести себя так, как будто он изолирован и по размерам больше, чем на самом деле.
http://xumuk.ru/kv/6.html
http://spravochnick.ru/himiya/kisloty_i_osnovaniya/teoreticheskoe_obosnovanie_principa_zhestkih_i_myagkih_kislot_i_osnovaniy/