1. Уравнение состояния газа Ван-дер Ваальса
Главная > Документ
| Информация о документе | |
| Дата добавления: | |
| Размер: | |
| Доступные форматы для скачивания: |
1. Уравнение состояния газа Ван-дер Ваальса
1. Реальный газ отличается от идеального.
Уравнение состояния идеального газа – приближенно:
1) М.В.Ломоносов еще в XVIII в. указывал, что давление, обусловленное ударами о стенку хаотически движущихся молекул, не будет подчиняться закону Бойля  (при
(при  ) при больших концентрациях молекул;
) при больших концентрациях молекул;
2) как мы знаем из теоремы Нернста, и при низких температурах  давление является функцией плотности
давление является функцией плотности  ;
;
3) существуют и другие количественные и качественные нарушения уравнения Клайперона-Менделеева  . В частности, реальные газы могут быть переведены в жидкое состояние, а идеальные, следуя этому уравнению, — нет.
. В частности, реальные газы могут быть переведены в жидкое состояние, а идеальные, следуя этому уравнению, — нет.
Основной причиной наблюдаемых отличий реальных газов от идеальных является наличие сил взаимодействия между молекулами реальных газов .
Силы взаимодействия можно классифицировать следующим образом:
а) химические или валентные, если возникло новое соединение;
б) кулоновские силы притяжения и отталкивания, если газ ионизирован
(плазма — квазинейтральный ионизированный газ);
в) молекулярные силы  силы Ван-дер-Ваальса (1837-1923).
силы Ван-дер-Ваальса (1837-1923).
Именно они нас и будут интересовать. Это силы притяжения между молекулами на “больших” расстояниях  диаметра молекул. Природа этих сил электростатическая, однако взаимодействие молекул не описывается законом Кулона, т.к. каждая молекула
диаметра молекул. Природа этих сил электростатическая, однако взаимодействие молекул не описывается законом Кулона, т.к. каждая молекула
( ядро и электронная оболочка) представляет собой электронейтральную систему.
Если в отсутствии внешних полей центры положительных и отрицательных зарядов не совпадают, то такие молекулы называются полярными.
Если электронная оболочка симметрична и центры положительных и отрицательных
зарядов в молекуле совпадают, то мы имеем дело с неполярными молекулами.
Под действием электрического поля
а) неполярные молекулы поляризуются за счет деформации электронной оболочки;
б ) полярные молекулы ориентируются по полю.
) полярные молекулы ориентируются по полю.
Вокруг каждой поляризованной молекулы возникает быстро убывающее с радиусом электрическое поле  .
.
Если в поле  молекулы 1 окажется молекула 2, то она будет поляризоваться и притягиваться к 1-ой молекуле под действием дисперсионной кулоновской силы.
молекулы 1 окажется молекула 2, то она будет поляризоваться и притягиваться к 1-ой молекуле под действием дисперсионной кулоновской силы.
П ри
ри 

 (т.к. это взаимодействие диполей, а не зарядов). Если электрические оболочки молекул перекрываются – проникают друг в друга, то силы притяжения переходят в силы отталкивания, которые экспоненциально убывают при .
(т.к. это взаимодействие диполей, а не зарядов). Если электрические оболочки молекул перекрываются – проникают друг в друга, то силы притяжения переходят в силы отталкивания, которые экспоненциально убывают при .
Энергия взаимодействия  двух молекул имеет следующий вид
двух молекул имеет следующий вид


 — потенциал Леннарда-Джонса является аппроксимацией энергии взаимодействия молекул
— потенциал Леннарда-Джонса является аппроксимацией энергии взаимодействия молекул  . При этом
. При этом
 — аппроксимация сил отталкивания,
— аппроксимация сил отталкивания,
 — аппроксимация сил притяжения.
— аппроксимация сил притяжения.
Аппроксимация (от лат. approximo–приближаюсь) –приближенное выражение какого-либо математическкого объекта через другие более простые.
Потенциал Леннарда-Джонса используется в теории газов и дает хорошее совпадение с экспериментом для реальных газов.
2. Модель: В простейшей теории используется ещё более грубая пунктирная аппроксимация,
соответствующая следующей модели:
1) Газ состоит из твердых упругих шариков диаметром  .
.
2) Молекулы-шарики притягиваются на расстоянии  радиуса сферы молекулярного действия
радиуса сферы молекулярного действия  при
при  .
.
3. Учёт притяжения.
Из-за взаимного притяжения между молекулами газ как бы сжимается эффективно большим давлением, чем давление  , оказываемое газом на стенки сосуда.
, оказываемое газом на стенки сосуда.
Величина этой положительной добавки к пропорциональна произведению числа
молекул, содержащихся в единице объема каждого из взаимодействующих объемов  и
и  , т.е.
, т.е.
 — квадрату концентрации:
— квадрату концентрации:
 — давление в реальном газе , — давление этого газа на стенки,
— давление в реальном газе , — давление этого газа на стенки,  — добавка, возникающая из-за наличия сил притяжения между молекулами.
— добавка, возникающая из-за наличия сил притяжения между молекулами.
4. Учет отталкивания.
Учтем силы отталкивания в рамках предложенной модели, предполагая существование объема недоступного для движения молекул вследствие конечности их размера .
П усть имеем всего две молекулы, одна покоится, а другая движется. Какой объем недоступен для движения второй молекулы?
усть имеем всего две молекулы, одна покоится, а другая движется. Какой объем недоступен для движения второй молекулы?
 , здесь
, здесь  — объем двух молекул.
— объем двух молекул.
Если у нас 1 моль, то объем, доступный движению молекул,  , где
, где  — несколько объемов молекул, содержащихся в моле.
— несколько объемов молекул, содержащихся в моле.
5. Уравнение Ван-дер-Ваальса.
Тогда подправим уравнение состояния одного моля идеального газа, написав
уравнение состояния для 1 моля газа Ван-дер-Ваальса:
 ,
,
где  , — постоянные коэффициенты, определенные для любого газа .
, — постоянные коэффициенты, определенные для любого газа .
Если у нас не один моль, а  молей, тогда
молей, тогда


 уравнение состояния газа Ван-дер-Ваальса .
уравнение состояния газа Ван-дер-Ваальса .
Еще раз о смысле каждой скобки,  и
и  : первая скобка – реальное давление внутри, вторая – объем, доступный движению, — притяжение молекул, — “мертвый” объем.
: первая скобка – реальное давление внутри, вторая – объем, доступный движению, — притяжение молекул, — “мертвый” объем.
6. Качественные вопросы
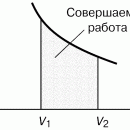
1) Изобразим изохору реального газа в координатах

То есть для реального газа увеличение температуры при постоянном объеме вызывает более резкий рост давления, чем для идеального газа:


 P
P 
 Т
Т 
2) Изобразим изобару в координатах 



T При 


 , т.е. второе слагаемое больше третьего
, т.е. второе слагаемое больше третьего
 график приближается к асимптоте сверху.
график приближается к асимптоте сверху.
2. Внутренняя энергия газа Ван-дер-Ваальса
1. Внутренняя энергия В-д-В газа 

 — потенциальное поле притяжения молекул
— потенциальное поле притяжения молекул
При расширении работа, совершаемая силами притяжения молекул газа, равна убыли его потенциальной энергии, связанной с притяжением молекул:

 — часть давления, обусловленная притяжением молекул.
— часть давления, обусловленная притяжением молекул.



2. Задача об убранной перегородке для реального газа .
а) Для идеального газа:


б) Для Ван-дер-Ваальсовского газа:


То есть с увеличением объема температура газа понижается, т.к. внутренняя энергия газа расходуется на работу против сил притяжения молекул. В этом отличие реального газа от идеального.
температура газа понижается, т.к. внутренняя энергия газа расходуется на работу против сил притяжения молекул. В этом отличие реального газа от идеального.
3. Эффект Джоуля-Томпсона
В опр. Что будет происходить с температурой реального газа после его прохождения через пористую перегородку, если
опр. Что будет происходить с температурой реального газа после его прохождения через пористую перегородку, если  ?
?
Отв. Для идеального газа температура не изменится, а для реального газа будет наблюдаться эффект Джоуля- Томпсона.
а) Если  , то эффект притяжения доминирует над отталкиванием, поэтому
, то эффект притяжения доминирует над отталкиванием, поэтому  , и температура газа понижается
, и температура газа понижается . Это — положительный эффект Джоуля-Томпсона.
. Это — положительный эффект Джоуля-Томпсона.
Газ охлаждается, так как при расширении внутренняя энергия расходуется на работу против сил молекулярного притяжения. Этот эффект используется для ожижения газов и получения низких температур.
б) При  и газ нагревается. Это – отрицательный эффект Джоуля-
и газ нагревается. Это – отрицательный эффект Джоуля-
Отрицательный эффект Джоуля-Томпсона реализуется для  и
и  , у которых
, у которых  , т.е. притяжение очень слабое, а свобода передвижения при расширении возрастает.
, т.е. притяжение очень слабое, а свобода передвижения при расширении возрастает.
Для общего развития:  ,
,
где 
 — энтальпия – еще один термодинамический потенциал.
— энтальпия – еще один термодинамический потенциал.

3. Теплоемкость газа Ван-дер Ваальса

Здесь  — это давление газа на стенку, так как работа совершается газом над внешними телами.
— это давление газа на стенку, так как работа совершается газом над внешними телами.

Проведем расчет теплоемкости для одного моля реального газа при различных процессах:


1) Изохорический 


 , как и для идеального газа
, как и для идеального газа
2) Адиабатический  политропические
политропические
 , как и для идеального газа процессы
, как и для идеального газа процессы
3)

4) 



Найдем  . Для этого необходимо найти
. Для этого необходимо найти  .
.




 ,
,
т.о.  , и изобарический процесс не является политропическим для газа
, и изобарический процесс не является политропическим для газа
4. Уравнение адиабаты для газа Ван-дер- Ваальса






 уравнение адиабаты В-д-В газа.
уравнение адиабаты В-д-В газа.
5. Энтропия газа Ван-дер Ваальса
1) 
 — энтропия одного моля.
— энтропия одного моля.
2) Как измениться энтропия газа в задаче об убранной перегородке:

То есть 
6. Уравнение политропы В-д-В газа
Уравнение адиабаты для В-д-В газа:


Для идеального газа уравнение адиабаты:


По аналогии естественно предположить, что для В-д-В газа уравнение политропы будет иметь вид:


 , так как процесс политропический
, так как процесс политропический  =
= .
.




или уравнения политропы В-д-В газа, здесь  .
.

7. Изотермы Ван-дер-Ваальса
 Т
Т
М ы уже изображали:
ы уже изображали:

в) Изотермы, построенные в координатах (P,V), дают наиболее содержательные результаты.

Найдем особые точки изотермы, вычислив  :
:





Найдем корни этого уравнения:


 точное решение дает
точное решение дает  , оно соответствует минимуму на изотерме В-д-В газа.
, оно соответствует минимуму на изотерме В-д-В газа.
 соответствует максимуму на изотерме В-д-В газа.
соответствует максимуму на изотерме В-д-В газа.
Т.о. чем  , тем ближе
, тем ближе  к
к  , и критическая изотерма имеет только точку перегиба К, которая называется критической точкой ( Эндрюс 1861-1869 ).
, и критическая изотерма имеет только точку перегиба К, которая называется критической точкой ( Эндрюс 1861-1869 ).
В точке К с параметрами  вещество находится в критическом состоянии.
вещество находится в критическом состоянии.
 — критическая температура (температура абсолютного кипения по Менделееву), выше этой температуры газ не может быть сконденсирован в жидкость никаким увеличением давления .
— критическая температура (температура абсолютного кипения по Менделееву), выше этой температуры газ не может быть сконденсирован в жидкость никаким увеличением давления .
К понятию критического состояния можно придти путем анализа экспериментальной изотермы без рассмотрения какого-либо теоретического уравнения состояния. Уравнение В-д-В является лишь модельным, и не все состояния, совместимые с уравнением В-д-В, могут быть реализованы. Реализованы могут быть только устойчивые состояния.
Одно из необходимых условий термодинамической устойчивости однородного вещества:

Оно означает, что при изотермическом увеличении давления  объем
объем  должен уменьшаться
должен уменьшаться . Следовательно, участок AB на изотерме должен быть выброшен.
. Следовательно, участок AB на изотерме должен быть выброшен.
Участки AC и BD соответствуют разным фазовым состояниям вещества.
8. Экспериментальные изотермы реального газа
В изучение этого вопроса значительный вклад внесли следующие исследователи: Ван Марум (1750-1837), Фарадей (1791-1867), Эндрюс (1813-1885).
Опред. В термодинамике фазой называется совокупность однородных, одинаковых по
свойствам частей системы .
Различают следующие фазы: газообразная, жидкая, различные кристаллические
Разные фазы могут существовать в равновесии друг с другом.
Переход вещества из одной фазы в другую называется фазовым переходом.
Существуют фазовые переходы:
жидкость  газ;
газ;
газ твердая фаза;
жидкость твердая фаза.
Фазовый переход I рода сопровождается выделением/поглощением тепла.
твердая фаза твердая фаза;
резистивное состояние сверхпроводящее состояние;
обычная жидкость сверхтекучая жидкость.
При фазовых переходах II рода нет выделения/поглощения тепла .
Р еальные изотермы имеют горизонтальный участок EF, на котором процесс сжатия не сопровождается постом вследствие образования другой фазы.
еальные изотермы имеют горизонтальный участок EF, на котором процесс сжатия не сопровождается постом вследствие образования другой фазы.
На участке EF жидкость и газ находятся в термодинамическом равновесии (т.д.р.)
Условия термодинамического равновесия фаз :

Таким образом мы распространили уравнение Ван-дер Вальса на область жидкого состояния, где оно вообще-то не применимо.
Газ, находящийся в равновесии с жидкостью, называется насыщенным паром .
Вопр.: Какая разница между паром и газом?
Отв. Пар – это газ, который может быть превращен в жидкость изотермически .
Пар – это газ с температурой ниже критической.
В значительной степени двойственная терминология обусловлена исторически: те газы, которые могли быть превращены в жидкость, называли парами, а те, которые ожижить не удавалось, — газами. Когда все газы сжижили, то необходимость в различии названий отпала.
В  критической т.К параметры системы
критической т.К параметры системы  можно определить по методу Эндрюса из серии экспериментальных изотерм, но это громоздко. Проще – метод исчезновения мениска. Обсудим изменение фазового состояния вещества (эфира) в запаянной стеклянной ампуле при изохорическом нагревании:
можно определить по методу Эндрюса из серии экспериментальных изотерм, но это громоздко. Проще – метод исчезновения мениска. Обсудим изменение фазового состояния вещества (эфира) в запаянной стеклянной ампуле при изохорическом нагревании:
Из т.1 в т.К: мениск остается на месте, но становится более горизонтальным, т.к. свойства пара и жидкости становятся более близкими. В т.К мениск исчезает.
Из т.2в т.Е (жидкости было «больше, чем нужно»): мениск повышается, пока вся мензурка не заполнится жидкостью.
Из т.3в т.F (жимдкости было «меньше, чем нужно»): мениск понижается, пока вся ампула не
На самом деле  — сжимаемость вещества в критической точке бесконечна,
— сжимаемость вещества в критической точке бесконечна,
следовательно, вещество внизу ампулы должно заметно уплотняться вследствие
гравитации, поэтому благодаря сильному изменению плотности вещества с высотой,
в ампуле иметь критическую плотность может только бесконечно тонкой слой вещества.
В нем и происходит исчезновение мениска. В этом заключается суть метода мениска для
измерения критической температуры  .
.
В критической точке вещество обладает необычными свойствами:
1) бесконечной сжимаемостью;
2) 
3 ) медленность установления равновесного состояния влечет за собой гистерезис плотности.
) медленность установления равновесного состояния влечет за собой гистерезис плотности.
Метастабильные участки AE и BF могут существовать:
BF соответствует пересыщенному пару, который может быть получен при резком адиабатическом расширении.
EA соответствует перегретой жидкости, она может быть получена резким уменьшением давления. Это – физически однородная жидкость, в которой нет растворенного газа.
Горизонтальный участок изотермы EF определяется правилом Максвелла:  .
.
Рассмотрим цикл  и применим к нему равенство Клаузиуса
и применим к нему равенство Клаузиуса  , так как процесс равновесный и это изотермы.
, так как процесс равновесный и это изотермы.

Вопр.: Рассмотрим два цикла  и
и  . Рассуждая аналогично, получим для любого из них:
. Рассуждая аналогично, получим для любого из них:  , то есть
, то есть  любого из них равна нулю – противоречие. В чем дело?
любого из них равна нулю – противоречие. В чем дело?
Отв.: 1) Для цикла в точке  происходит переход однофазного вещества в двухфазное, а это необратимый процесс
происходит переход однофазного вещества в двухфазное, а это необратимый процесс  , что и существует на самом деле.
, что и существует на самом деле.
2) Для цикла ,  — такой цикл невозможен, так как в точке двухфазное
— такой цикл невозможен, так как в точке двухфазное
состояние не может перейти в однофазное. В обратную сторону перход возможен, но при этом  и противоречия нет.
и противоречия нет.
К ак соотносятся
ак соотносятся  и
и  в точке ?
в точке ?
 — правило рычага
— правило рычага
9. Уравнение Клайперона-Клаузиуса. Диаграмма состояния

1234 – цикл Карно с изотермическим реальным газом.


 — уравнение Клайперона-Клаузиуса.
— уравнение Клайперона-Клаузиуса.
Тангенс угла наклона кривой испарения и
сублимации всегда больше нуля,
следовательно, угол наклона соответствующих кривых меньше  .
.
Для плавления это может быть не так ( ).
).

Диаграмма фазовых переходов 
Три фазы одного и того же вещества могут существовать в равновесии только в
одной точке, то есть при единственных значениях  . Эта точка называется тройной.
. Эта точка называется тройной.
Р авновесие более, чем трех фаз одного и того же вещества невозможно.
авновесие более, чем трех фаз одного и того же вещества невозможно.
Параметры критичного состояния для :  , 218 атм,
, 218 атм,  .
.
10. Другие уравнения реальных газов
 (при умеренных давлениях лучше согласуется с опытом)
(при умеренных давлениях лучше согласуется с опытом)
 (точнее за счет лишнего подгоночного параметра)
(точнее за счет лишнего подгоночного параметра)
 , где
, где  — вариальные коэффициенты.
— вариальные коэффициенты.
Уравнение Ван-дер Вальса имеет преимущества вследствие легко объяснимого физического смысла параметров. Его-то и надо уметь пояснять.
ВАН-ДЕР-ВААЛЬСА УРАВНЕНИЕ
ВАН-ДЕР-ВААЛЬСА УРАВНЕНИЕ – модельное уравнение состояния реального газа, учитывающее, в отличие от уравнения состояния идеального газа, взаимодействие молекул между собой, а именно: мощное отталкивание на малых расстояниях R между центрами масс молекул

(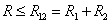 ) и их притяжение на больших
) и их притяжение на больших
(R > R12) расстояниях. Здесь R1 и R2 – газокинетические радиусы молекул. В ряде случаев, для простоты, используется средний газокинетический диаметр взаимодействующих молекул 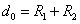 , очевидно для одинаковых молекул
, очевидно для одинаковых молекул 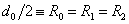 .
.
Уравнение состояния является функциональной связью между четырьмя термодинамическими параметрами состояния физической системы. Для описания однокомпонентных (состоящих из частиц одного сорта) физических систем достаточно четырех параметров. Для систем, состоящих из различных частиц (например, воздух – смесь азота, кислорода, аргона, углекислого газа и др.), полный перечень необходимых параметров включает относительные концентрации компонент смеси. Для простоты, будут рассмотрены только однокомпонентные системы. Традиционный и наиболее употребительный набор параметров состояния состоит из массы системы m, давления p, объема V и температуры T. Использование массы системы в качестве одного из ее параметров предполагает, что известна молярная масса вещества 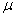 , из которого она состоит. Набор параметров состояния
, из которого она состоит. Набор параметров состояния 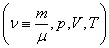 «продиктован» экспериментом, так как все входящие в него величины достаточно просто и непосредственно измеряются. Здесь
«продиктован» экспериментом, так как все входящие в него величины достаточно просто и непосредственно измеряются. Здесь 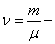 число молей. Разумеется, возможны и другие наборы параметров состояния: число частиц в системе
число молей. Разумеется, возможны и другие наборы параметров состояния: число частиц в системе 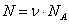 , объем, энтропия и внутренняя энергия (NA – число Авогадро).
, объем, энтропия и внутренняя энергия (NA – число Авогадро).
Уравнение состояния идеального газа (газа невзаимодействующих материальных точек) было получено Э.Клапейроном (1834) в результате объединения трех экспериментально установленных газовых законов: 1) Р.Бойля (1662) и Э.Мариотта (1676); 2) Шарля (1785); 3) Гей-Люссака (1802). Сейчас это уравнение (здесь R – универсальная газовая постоянная)
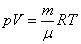
называют уравнением Клапейрона – Менделеева.
В данном частном случае заслуга Д.И.Менделеева в том, что он вывел написанное выше универсальное уравнение состояния идеальных газов. В частности, при исследовании явлений, не укладывающихся в модель идеального газа и обусловленных взаимодействием молекул между собой (поверхностное натяжение жидкостей и сопутствующие капиллярные явления, непрерывные и скачкообразные фазовые переходы жидкость – газ), Менделеев ввел понятие температуры «абсолютного» кипения, которая впоследствии была названа Эндрюсом критической температурой – температурой критического состояния вещества, это уже сфера непосредственных применений уравнения Ван-дер-Ваальса.
Учет взаимодействия между молекулами газа при расчете его термодинамических характеристик впервые был выполнен в 1873 голландским физиком Я.Д.Ван-дер-Ваальсом, именем которого названо полученное им уравнение состояния такого газа. Строго говоря, ван-дер-ваальсовским можно называть газ, потенциальная энергия притяжения молекул которого на больших расстояниях убывает с ростом R по закону
его, например, нет места в плазме состоящей из заряженных частиц, потенциальная энергия взаимодействия которых на больших расстояниях убывает в соответствии с законом Кулона
т.е существенно медленнее.
Силы Ван-дер-Ваальса (R > d0)
для молекулярных и атомарных газов носят достаточно универсальный характер. Квантовомеханическое усреднение потенциальной энергии по взаимным ориентациям взаимодействующих объектов практически во всех случаях приводит к асимптотическому закону (1), (3).
Во-первых, это взаимодействие полярных молекул, т.е. молекул с собственным электрическим дипольным моментом (молекулы типа HCl, H2O и т.п.). Соответствующие силы называют ориентационными.
Во-вторых, взаимодействие полярной и неполярной молекулы (не имеющей собственного электрического дипольного момента): He, Ar, … N2, O2 … . Такое взаимодействие принято называть индукционным.
Наконец, взаимодействие неполярных атомов и молекул – дисперсионное взаимодействие. Происхождение дисперсионных сил строго объясняется только в рамках квантовой механики. Качественно возникновение этих сил можно объяснить – в результате квантовомеханических флуктуаций у неполярной молекулы возникает мгновенный дипольный момент, его электрическое поле поляризует другую неполярную молекулу и у неё появляется наведенный мгновенный дипольный момент. Энергия взаимодействия неполярных молекул – это квантовомеханическое среднее энергии взаимодействия таких мгновенных диполей. Дисперсионные силы не зависят от наличия или отсутствия собственных дипольных моментов у атомов и молекул и потому всегда имеют место. В случае неполярных атомов и молекул дисперсионные силы в десятки и даже сотни раз больше сил ориентационных и индукционных. В случае молекул с большим собственным дипольным моментом, например, молекул воды H2O, дисперсионная сила в три раза меньше ориентационной. Все эти силы имеют асимптотику (3), таким образом, в общем случае усредненная потенциальная энергия
(4) 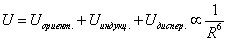 при
при 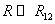 .
.
Мощное отталкивание молекул на малых расстояниях возникает при перекрытии внешних заполненных электронных оболочек и обусловлено принципом запрета Паули. Зависимость этих сил от R нельзя объяснить в рамках чисто классической электродинамики. Силы отталкивания в большей мере, чем силы притяжения, зависят от конкретных особенностей строения электронных оболочек взаимодействующих молекул и требуют для своего определения громоздких квантовомеханических расчетов. Хорошее согласие с экспериментом дает следующая модель
Из (5) видно, что уменьшение расстояния в два раза приводит к увеличению силы отталкивания 15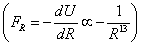 более чем в 8 тысяч раз, что и позволяет говорить о «мощных» силах отталкивания.
более чем в 8 тысяч раз, что и позволяет говорить о «мощных» силах отталкивания.
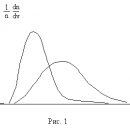
При практических расчетах широко используется модельный потенциал Ленард – Джонса, (с учетом (1) и (5))
показанный на рис. 1. Видно, что параметр D имеет смысл глубины потенциальной ямы, а параметр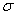 определяет ее размер: абсцисса минимума
определяет ее размер: абсцисса минимума 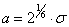 .
.
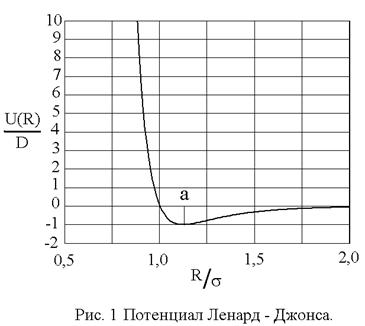
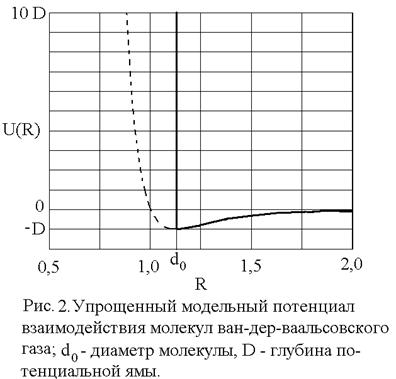
Уравнение состояния ван-дер-ваальсовского газа, само по себе приближенное, может быть, тем не менее, точно получено в рамках модели притягивающихся твердых шаров. В этой модели весьма большие, но конечные силы отталкивания на малых расстояниях заменяются бесконечно большими силами, что означает замену близкого к вертикали криволинейного потенциального барьера левее точки минимума (рис. 1) вертикальной потенциальной стенкой в соответствующей точке: R = d0, что показано на рис. 2. При расстояниях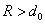 сохраняется зависимость от R по формуле (6).
сохраняется зависимость от R по формуле (6).
Вертикальная потенциальная стенка ставится именно в точке R = d0 = 2R0, т.к. минимальное расстояние между центрами двух твердых шаров равно их диаметру.
Притяжение молекул на расстояниях дает поправку к внутренней энергии газа, равную энергии их взаимодействия: Uвз. При достаточной разреженности газа с хорошей точностью справедливо предположение о попарном взаимодействии молекул, что приводит к выражению для Uвз:
дает поправку к внутренней энергии газа, равную энергии их взаимодействия: Uвз. При достаточной разреженности газа с хорошей точностью справедливо предположение о попарном взаимодействии молекул, что приводит к выражению для Uвз:
(7) 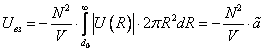 , 24
, 24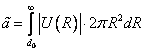
Где 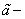 одна из двух постоянных Ван-дер-Ваальса, учитывающая притяжение между молекулами, N – число молекул в газе, V – объем газа. Можно просто объяснить аналитическую структуру выражения (7). Число молекул в единице объема
одна из двух постоянных Ван-дер-Ваальса, учитывающая притяжение между молекулами, N – число молекул в газе, V – объем газа. Можно просто объяснить аналитическую структуру выражения (7). Число молекул в единице объема 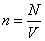 , взаимодействуют они попарно, число пар молекул в единице объема
, взаимодействуют они попарно, число пар молекул в единице объема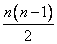 , что, учитывая макроскопичность n, можно считать точно равным
, что, учитывая макроскопичность n, можно считать точно равным 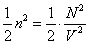 . Средняя энергия взаимодействия одной пары молекул, как видно из структуры интеграла в (7), равна
. Средняя энергия взаимодействия одной пары молекул, как видно из структуры интеграла в (7), равна 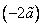 , откуда для объема V, в силу аддитивности энергии, получается
, откуда для объема V, в силу аддитивности энергии, получается 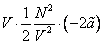 , т.е. формула (7).
, т.е. формула (7).
Внутренняя энергия реального газа складывается из суммарной кинетической энергии его молекул (той же, что и в идеальном газе при тех же параметрах состояния) и потенциальной энергии их взаимодействия. Отсюда, например, для одноатомного газа с температурой T можно записать:
Конечность объема молекул приводит к тому, что не весь объем сосуда V доступен для их движения – уменьшается «свобода» размещения молекул газа в его фазовом пространстве, что, в свою очередь, уменьшает статистический вес макросостояния и энтропию газа. Энтропия идеального (молекулы – материальные точки) одноатомного газа с температурой , занимающего сосуд объемом V, имеет вид
Если объем недоступный для движения молекул – шариков реального газа, равен V0, то его энтропия
Для двух молекул радиуса R0 с минимальным расстоянием между центрами 2R0, объем, недоступный для движения, – это объем сферы, равный
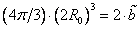 , где
, где 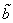 – учетверенный объем одной молекулы, это вторая константа Ван-дер-Ваальса.
– учетверенный объем одной молекулы, это вторая константа Ван-дер-Ваальса.
Недоступный для движения объем в расчете на одну молекулу равен 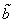 , а для N молекул –
, а для N молекул –
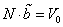 , откуда окончательное выражение для энтропии одноатомного ван-дер-ваальсовского газа имеет вид
, откуда окончательное выражение для энтропии одноатомного ван-дер-ваальсовского газа имеет вид
В рамках рассматриваемой модели параметры а и b (вторые формулы в (8) и (12)) являются атомными константами (диаметр молекулы d0 считается фиксированной величиной, не зависящей от температуры, хотя, строго говоря, это не так), не зависящими параметров термодинамического состояния вещества.
Основное термодинамическое тождество имеет вид
это первое начало термодинамики, в которое для квазистатических процессов подставлены выражения для получаемой системой теплоты и (–pdV) для совершаемой над системой работы, оно позволяет получить уравнение состояния Ван-дер-ваальсовского газа с выражения для давления, следующего из (12)
В (13) индекс S указывает на то, что дифференцировать нужно при постоянной энтропии. Подстановка (8) и (11) в (13) приводит к уравнению состояния реального газа Ван-дер-Ваальса
Переход от числа молекул в газе N к числу молей 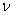 осуществляется с помощью замены
осуществляется с помощью замены 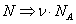 , где NA – число Авогадро и соответствующего этой замене переопределения постоянных Ван-дер-Ваальса
, где NA – число Авогадро и соответствующего этой замене переопределения постоянных Ван-дер-Ваальса
В этих переменных уравнение Ван-дер-Ваальса имеет вид 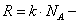 ( универсальная газовая постоянная):
( универсальная газовая постоянная):
Главное значение уравнения Ван-дер-Ваальса состоит, во-первых, в простоте и физической понятности его аналитической структуры: поправка a учитывает притяжение молекул на больших расстояниях, поправка b – их отталкивание на малых расстояниях. Уравнение состояния идеального газа 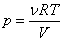 получается из (16) путем предельного перехода a → 0, b → 0. стрелки
получается из (16) путем предельного перехода a → 0, b → 0. стрелки
Во-вторых, уравнение Ван-дер-Ваальса обладает (несмотря на приближенность модели) широким спектром качественных, а в ряде случаев и полуколичественных предсказаний о поведении реального вещества, которые следуют из анализа уравнения (16) и вида соответствующих ему изотерм и касаются поведения вещества не только в достаточно разреженном газообразном состоянии, но и в жидком и двухфазном состояниях, т.е. в состояниях, далеких от априорной области применимости модели Ван-дер-Ваальса.
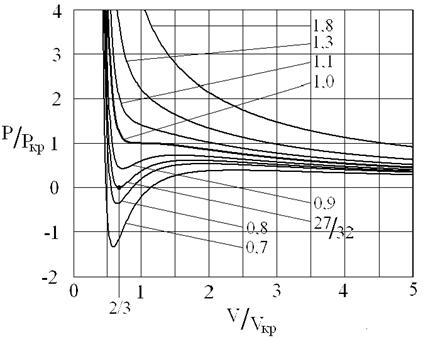
Рис. 3. Изотермы Ван-дер-Ваальса. Цифры, указывают отношение температуры, соответствующей данной изотерме, к критической температуре вещества. Единица соответствует критической изотерме T = Tкр.
Уравнение (16) имеет особую точку – точку перегиба, в которой
это соответствует реальной физической особенности – критическому состоянию вещества, в котором исчезает различие между жидкостью и ее паром (жидкой и газовой фазами), находящимися в состоянии термодинамического равновесия. Критическая точка является одним из концов кривой равновесия жидкость – пар на диаграмме (p, T), другим концом этой кривой является тройная точка, в которой в термодинамическом равновесии находятся все три фазы: газовая, жидкая и кристаллическая. Критической точке соответствуют критическая температура Tкр., критическое давление pкр. и критический объем Vкр. При температурах выше критической переход «жидкость – пар» происходит без скачка плотности, в критической точке исчезает мениск в капилляре, обращается в нуль теплота испарения и в бесконечность изотермическая сжимаемость (пропорциональная производной 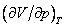 ).
).
Решение уравнений (17) дает связь критических параметров с постоянными Ван-дер-Ваальса a и b:
Формулы (18) позволяют найти константы а и b по экспериментально определенным параметрам критического состояния. Одним из показателей количественной точности уравнения Ван-дер-Ваальса является результат критического коэффициента 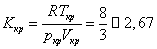 , следующего из (18) c его экспериментальным значением
, следующего из (18) c его экспериментальным значением
| Вещество | Kкр, эксперимент | Вещество | Kкр, эксперимент |
| H2 | 3,03 | SO2 | 3,60 |
| He | 3,13 | C6H6 | 3,76 |
| N2 | 3,42 | H2O | 4,46 |
| O2 | 3,42 | CO2 | 4,49 |
Если для водорода 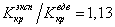 , то для углекислого газа это отношение уже составляет
, то для углекислого газа это отношение уже составляет 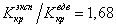 .
.
Критическое давление и критическая температура, например, воды равны соответственно: 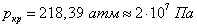 ,
, 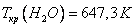 .
.
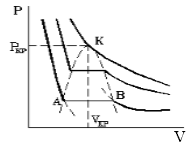
На рис. 4 дано несколько утрированное (по сравнению с рис. 3) качественное изображение изотермы Ван-дер-Ваальса с температурой ниже критической. На участке 3–4–5 производная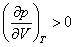 , что нереализуемо. Вещество в таком состоянии существовать не может, так как оно термодинамически неустойчиво: увеличение объема при постоянной температуре приводит к увеличению давления. Невозможность существования вещества в таком состоянии вытекает и из второго начала термодинамики, из которого строго следует, что для любого вещества изотермическая производная
, что нереализуемо. Вещество в таком состоянии существовать не может, так как оно термодинамически неустойчиво: увеличение объема при постоянной температуре приводит к увеличению давления. Невозможность существования вещества в таком состоянии вытекает и из второго начала термодинамики, из которого строго следует, что для любого вещества изотермическая производная 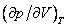 может быть только меньше нуля, это плата за интерполяционный характер модели Ван-дер-Ваальса. Поправка к энергии рассчитана в предположении парности
может быть только меньше нуля, это плата за интерполяционный характер модели Ван-дер-Ваальса. Поправка к энергии рассчитана в предположении парности
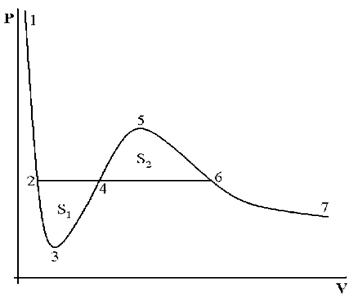
взаимодействия молекул, что справедливо при малых плотностях (больших объемах) – это «хвост» 6–7 изотермы на рис. 4, где она асимптотически сближается с изотермой идеального газа. Поправка на конечность размера молекул сделана в предположении, что молекулы ведут себя как твердые шары, это приближенно имеет место в жидкости при объемах, близких к суммарному объему плотно упакованных молекул, чему соответствует участок 1–2 изотермы. Следовательно, только на «концах» 1–2 и 6–7 изотермы ван-дер-ваальсовский подход может претендовать на приближенное количественное и правильное качественное описание. В промежуточной области теоретических оснований для таких претензий нет и, как следствие, получается нефизический участок 3–4–5.
Таким образом, участок 1–2 может быть поставлен в соответствие жидкому состоянию вещества. Его вертикальность обусловлена тем, что при V стрелка vb знаменатель в первом слагаемом (16) обращается в нуль и кривая выходит на вертикальную асимптоту V = vb = V0. В эксперименте этому соответствует ничтожно малая (по сравнению с газами) сжимаемость жидкостей. На участке 6–7 изотерма Ван-дер-Ваальса близка к изотерме идеального газа.
При температурах ниже критической, когда только и есть нефизический участок 3–4–5 (см. рис. 3), переход из жидкого состояния 1–2 в газовое состояние 6–7 происходит только через двухфазное состояние. Известно, что давление в двухфазной системе есть однозначная функция температуры, поэтому в переменных (p, V) соответствующий двухфазному состоянию На рис. 4 участок изотермы является отрезком горизонтальной прямой (участок 2–4–6 на рис. 4).
В уравнении Ван-дер-Ваальса нет горизонтального участка, однако наличие ван-дер-ваальсовского нефизического участка 3–4–5 позволяет теоретически определить «высоту» двухфазного участка 2–4–6, т.е. давление в двухфазной системе при данной температуре.
Если рассмотреть циклический процесс 2–3–4–5–6–4–2 (замкнутый контур ) и, учитывая, что во всех точках этого цикла температура постоянна, проинтегрировать основное термодинамическое тождество, то получим
(19) . 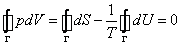
Равенство нулю интегралов в правой части (19) есть следствие замкнутости процесса и того, что энтропия S и внутренняя энергия U – функции состояния. Равенство нулю интеграла 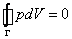 означает, что двухфазный участок следует расположить так, чтобы площади S1 и S2 (рис. 4) были равны (правило Максвелла).
означает, что двухфазный участок следует расположить так, чтобы площади S1 и S2 (рис. 4) были равны (правило Максвелла).
Участки 2–3 и 5–6 соответствуют реальным метастабильным состояниям вещества, а именно: 2–3 – перегретая жидкость, 6–5 – переохлажденный (пересыщенный) пар. В этих состояниях жидкость или пар могут существовать в течение некоторого времени, если нет центров парообразования и конденсации. Появление в жидкости центров парообразования ведет к немедленному возникновению и росту на их месте пузырьков пара. Аналогично, появление центров конденсации в переохлажденном паре ведет к немедленному возникновению и росту на их месте капель жидкости. Оба явления используются для регистрации треков заряженных частиц: первое в пузырьковой камере, второе в камере Вильсона (туманной камере). Роль центров парообразования (конденсации) играют ионы, которые оставляет на своем пути заряженная частица в результате ионизации молекул жидкости (пара) при столкновениях с ними. Пузырьки (капли) существуют достаточное для их фотографирования время, что делает видимой траекторию, по которой двигалась заряженная частица. Исследование трека частицы позволяет определить ее энергию и импульс, соответственно, вычислить ее массу, что является одной из важнейших задач физики элементарных частиц.
При температуре 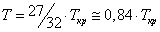 , что для воды составляет 273° C, минимум ван-дер-ваальсовской изотермы достигает нуля давления. При более низких температурах (рис. 3, кривые 0,8 и 0,7) давление в окрестности минимума становится отрицательным, что означает, что жидкость из-за действия сил притяжения между ее молекулами может «сопротивляться растяжению» (подобно пружине). Растянутую жидкость (например, ртуть) можно получить экспериментально, беря запаянную с одного конца стеклянную трубку длиной около метра и погружая ее в горизонтальную кювету с ртутью. После заполнения трубки ртутью трубку медленно, без встряхиваний поднимают в вертикальное положение, при этом в трубке наблюдается столб ртути, длина которого заметно превышает длину, соответствующую внешнему давлению, например, 760 мм.
, что для воды составляет 273° C, минимум ван-дер-ваальсовской изотермы достигает нуля давления. При более низких температурах (рис. 3, кривые 0,8 и 0,7) давление в окрестности минимума становится отрицательным, что означает, что жидкость из-за действия сил притяжения между ее молекулами может «сопротивляться растяжению» (подобно пружине). Растянутую жидкость (например, ртуть) можно получить экспериментально, беря запаянную с одного конца стеклянную трубку длиной около метра и погружая ее в горизонтальную кювету с ртутью. После заполнения трубки ртутью трубку медленно, без встряхиваний поднимают в вертикальное положение, при этом в трубке наблюдается столб ртути, длина которого заметно превышает длину, соответствующую внешнему давлению, например, 760 мм.
Савельев И.В. Курс общей физики, т. 3, Молекулярная физика и термодинамика, М.: Наука, Физматлит, 1998;
Сивухин Д.В. Общий курс физики, т. 2, Термодинамика и молекулярная физика, М., Физматлит, 2003;
Вдовиченко Н.В. Развитие фундаментальных принципов статистической физики в первой половине XX века. М., НАУКА, 1986.
Уравнение Ван-дер-Ваальса
Вы будете перенаправлены на Автор24
Что такое реальный газ
Реальным газом называют газ, между молекулами которого существуют заметные силы взаимодействия. В неидеальных, газах под высоким давлением, газах с большой плотностью взаимодействие молекул велико и его необходимо учитывать. Силы притяжения играют наиболее существенную роль на больших расстояниях между молекулами. Расстояние уменьшается, силы притяжения растут, но до определенного предела, затем они начинают уменьшаться и переходят в силы отталкивания. Притяжение и отталкивание молекул можно разделить и рассматривать и учитывать отдельно друг от друга.
Уравнение Ван-дер-Ваальса
Уравнение Ван-дер-Ваальса, описывающее состояние 1 моля реального газа, имеет вид:
где d- диаметр молекулы,
величина a вычисляется по формуле:
где $W_p\left(r\right)$- потенциальная энергия притяжения двух молекул. Необходимо заметить, что газовая постоянная имеет индивидуальное значение для каждого вещества. Она отличается от молярной газовой постоянной, причем она меньше, что говорит об объединении молекул вещества в комплексы около критического состояния. Вдали от критических состояний можно использовать универсальную газовую постоянную.
С увеличением объема роль поправок в уравнении (1) становится менее существенной. И в пределе уравнение (1) переходит в уравнение состояния идеального газа для 1 моля (4):
Уравнение (4) — уравнение Менделеева — Клайперона, где m- масса газа, $R=8,31\ \frac<Дж><моль\cdot К>$- универсальная газовая постоянная.
Это согласуется с тем фактом, что при уменьшении плотности реальные газы по своим свойствам приближаются к идеальным.
Уравнение (1) может быть записано в вириальной форме:
Для анализа изотерм уравнение (1) удобнее представить в виде:
Рассматриваемое уравнение может описывать и свойства жидкости, например плохую ее сжимаемость.
На рис.1 изображена изотерма Ван-дер-Ваальса для некоторого постоянного значения температуры T, построенная из соответствующего уравнения.
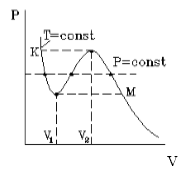
Такая зависимость на практике невозможна. Опыт показывает, что график должен иметь вид рис.2 то есть существуют области, в которых при изменении объема давление неизменно. В некоторых отрезках график изотермы параллелен оси V (рис 2). Это область фазового перехода. Жидкость и газ существую одновременно.
По мере увеличения температуры участок, отражающий состояние нахождения газа одновременно в двух фазах на графиках p(V), сужается и превращается в точку (рис. 2). Это особая точка К, в которой пропадает различие между жидкостью и паром. Это так называемая критическая точка.
Готовые работы на аналогичную тему
Итак, уравнение Ван-дер-Ваальса описывает поведение газов близких к реальным. Их можно применить к газообразной и жидкой фазам вещества. Эти уравнения отражают существование фазового перехода газ — жидкость. Показывают наличие критической точки перехода. Однако абсолютно точных количественных результатов расчеты, в которых используются вышеназванные уравнения, не дают.
Задание: Газ в количестве 1 моль находится в сосуде объемом V л при температуре $T_1$ давление газа $p_1$, а при $T_2$ давление газа $p_2$. Найти постоянные Ван-дер-Ваальса.
Запишем уравнение Ван-дер-Ваальса для одного моля реального газа для состояний 1 и 2:
Раскроем скобки в (1.1):
Вычтем $\left(1.4\right).\ из\ \left(1.3\right):$
Выразим a из (1.1):
Задание: Для реального газа, используя уравнение Ван-дер-Ваальса, получите уравнение адиабаты в параметрах V и T.
\[\delta Q=dU+\partial A=0\ \left(\ 2.1\right)\]
Так как процесс адиабатный, то он идет теплообмена. Перепишем уравнение (2.1) для ван-дер-ваальсовского газа, зная, что:
Из уравнения Ван-дер-Ваальса:
Подставим (2.5) в (2.4), разделим переменные:
Ответ: Уравнение адиабаты для заданного случая имеет вид: $<\left(V-b\right)>^<\frac<2>\nu >T=const.$
Получи деньги за свои студенческие работы
Курсовые, рефераты или другие работы
Автор этой статьи Дата последнего обновления статьи: 15 01 2022
http://www.krugosvet.ru/enc/fizika/van-der-vaalsa-uravnenie
http://spravochnick.ru/fizika/molekulyarnaya_fizika/uravnenie_van-der-vaalsa/