Урок 21. Теплота образования
В уроке 21 «Теплота образования» из курса «Химия для чайников» рассмотрим что такое теплоты образования и откуда их брать; выясним, какие условия считаются стандартными, а также различия простых и сложных веществ; для закрепления полученных знаний решим пару задач, где используются теплоты образования и закон аддитивности теплот реакций. Будет неплохо, если перед прочтением данного урока, вы изучите материал про изменение энтальпии, а также про теплоты сгорания.

Благодаря закону Гесса (закону аддитивности теплот реакций) нам совсем не нужно считать теплоты всех реакций; достаточно иметь сведения о теплОтах лишь того минимума реакций, из которых можно получить все остальные. Подобный минимум, принятый всеми учеными и инженерами, представляет собой теплоты образования соединений из входящих в них чистых элементов в стандартных состояниях . А что же такое: стандартные состояния?
Стандартное состояние вещества
Для кристаллических и жидких веществ стандартное состояние определяется как наиболее распространенная форма элемента при 25°С (298 К*) и внешнем давлении 1 атмосфера (атм); стандартное состояние газов определяется аналогичным образом, но при парциальном ** давлении 1 атм. Вот например, стандартным состоянием углерода считается графит, а не алмаз.
* Кельвин (обозначение «К») – еще один показатель измерения температуры, наряду с Цельсия. Пока вам просто следует запомнить, что 0°С = 273 К, соответственно 1°С = 274 К, -1°С = 272 К, а -273°С = 0 К. Самой низкой температурой считается 0 К и называется абсолютным нулем .
** Парциальное давление — это давление, которое имел бы газ, входящий в состав газовой смеси, если бы он один занимал объём, равный объёму смеси при той же температуре.
Простые и сложные вещества
Все вещества можно разделить на две обширные группы — простые и сложные вещества.
Простые вещества — это вещества, образованные из атомов одного элемента. Примеры простых веществ: молекулярные (O2, O3, H2, Cl2) и атомарные (He, Ar) газы; различные формы углерода, иод (I2), металлы (не в виде сплавов).
Сложные вещества (химические соединения) — это вещества, образованные атомами разных элементов. Так, оксид меди CuO образован атомами элементов меди Cu и кислорода O, вода H2O — атомами элементов водорода H и кислорода O.
Теперь вы понимаете различия простых веществ от сложных. Так вот, теплота образования простых веществ всегда равна нулю. Это, пожалуйста, запомните.
Стандартные теплоты образования
Вспомним прошлый урок, где мы пытались синтезировать алмаз. Теплоты образования всех веществ, участвующих в этом синтезе таковы:
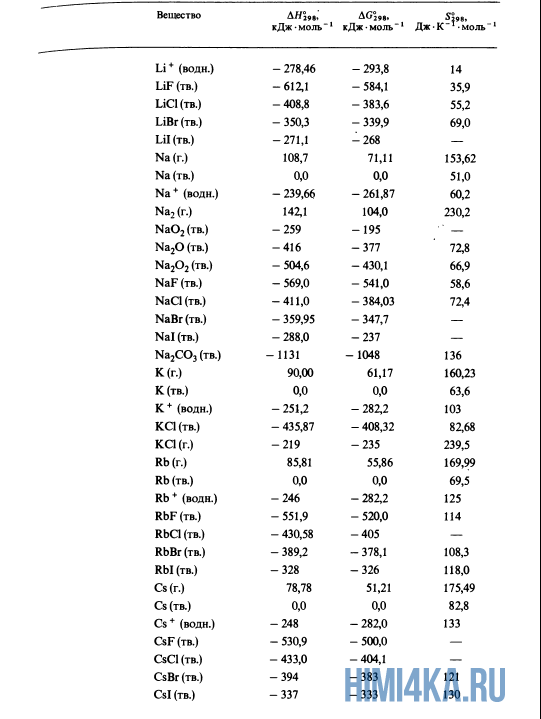
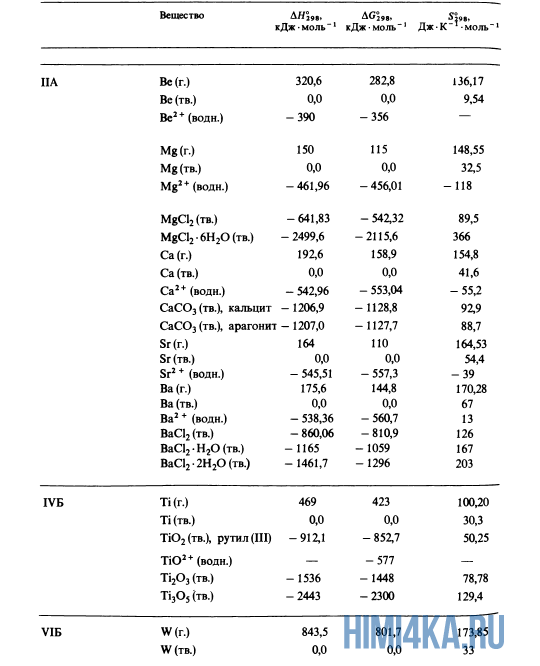
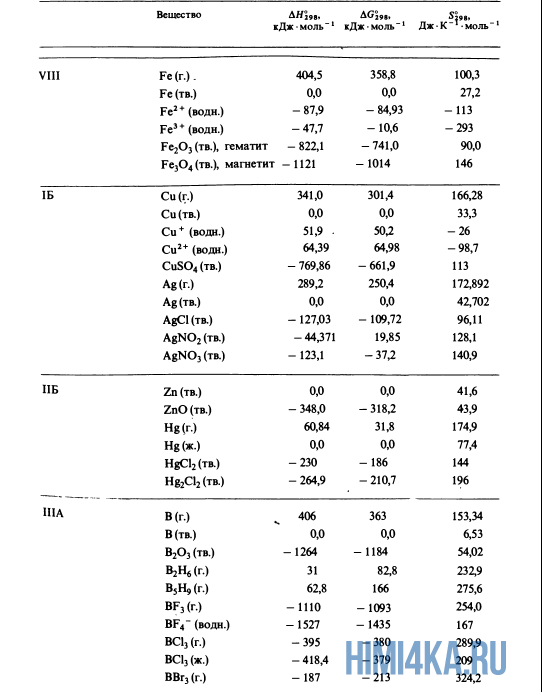
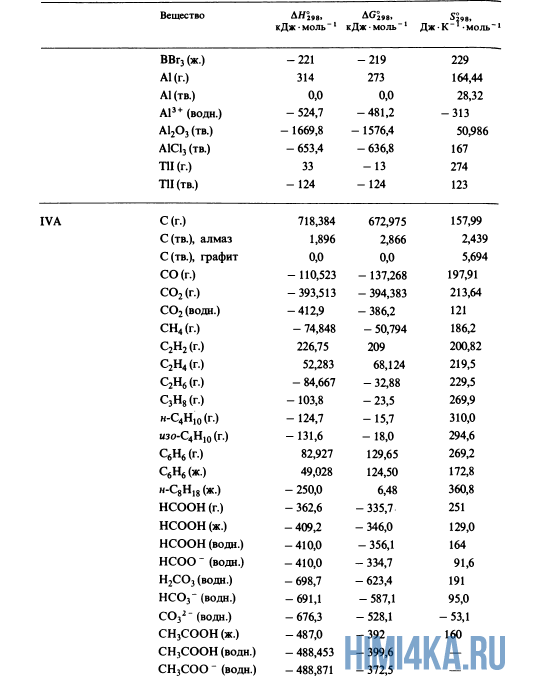
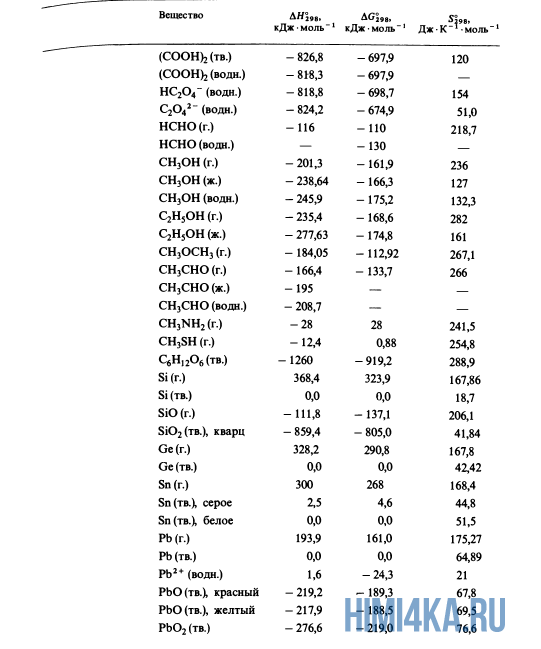
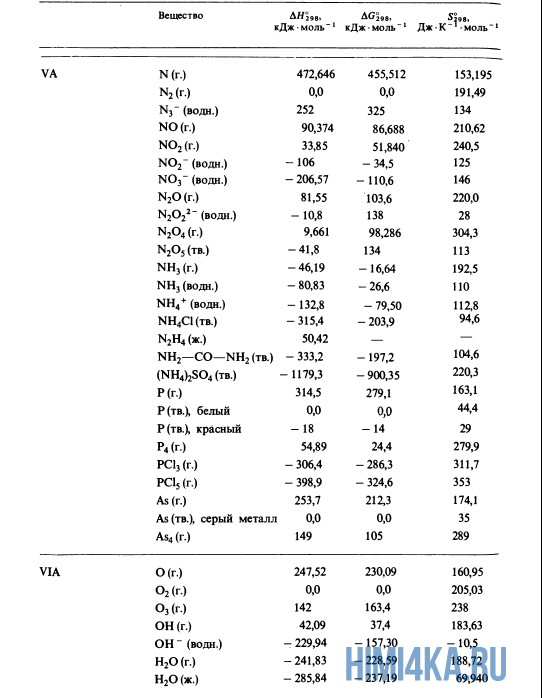
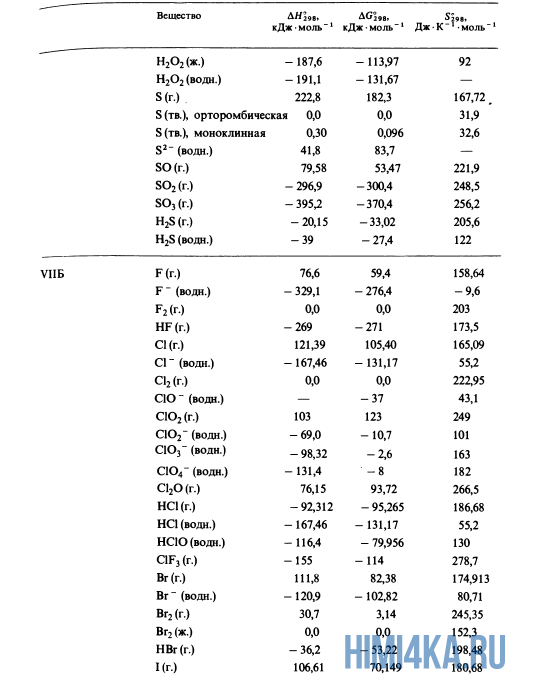
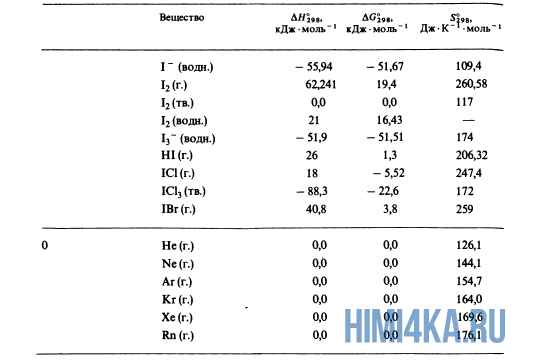
Как уже было отмечено выше, O2 относится к простым веществам, поэтому его теплота образования равна нулю. Таблицы стандартных теплот образования ΔH°298 соединений из чистых элементов приведена ниже под спойлером. В этой таблице нижний индекс 298 обозначает комнатную температуру (298 К = 25°С), а верхний индекс (знак градуса) означает, что реагенты и продукты находятся в своих стандартных состояниях. Однако не обязательно пользоваться записью ΔH°298, достаточно лишь написать ΔH.

Сейчас, на примере старой и доброй реакции (4) получения алмаза путем окисления метана, я покажу вам, как по теплотам образования веществ можно определить теплоту общей реакции. Это уравнение можно получить суммированием уравнения (2) с удвоенным уравнением (3) и обращенным уравнением (1):
В итоге получаем:
Теплота реакции вычисляется точно таким же способом, просто из теплот образования продуктов реакции вычитаем теплоты образования реагентов , учитывая все коэффициенты:
У кого возник вопрос, почему же мы не вычитаем теплоту образования кислорода, ответ ищите в таблице теплот образования.
Очевидно, что при таких вычислениях следует быть очень внимательным к знакам и коэффициентам. Чтобы не допустить ошибки, настоятельно рекомендую вам выписывать отдельно каждое уравнение с соответствующей теплотой реакции в таком виде, чтобы сумма всех индивидуальных уравнений давала требуемую реакцию. Если все коэффициенты какого-либо уравнения умножены на произвольное число n, на это же число n должна быть умножена соответствующая теплота образования, а если используется обращенное уравнение реакции образования, то следует изменить свой знак перед табличной величиной ΔH. Когда индивидуальные уравнения суммируются для получения требуемой реакции, сумма индивидуальных теплот образования дает искомую теплоту реакции. А теперь закрепим полученные знания примерами.
Пример 1. Чему равна стандартная теплота реакции восстановления оксида трехвалентного железа Fe2O3 углеродом С до железа Fe и моноксида углерода O, протекающей в доменной печи?
Запишем уравнение реакции, указав под каждым веществом его стандартную теплоту образования:
Стандартная теплота образования простых элементов (С и Fe) по определению равна нулю. Стандартная теплота рассматриваемой реакции равна:
Как видите, изменение энтальпии положительно, а значит реакция эндотермическая (поглощается тепло). Полученный результат согласуется с известным фактом, что при восстановлении железной руды до свободного железа необходимо подводить к реакционной системе большое количество теплоты. Отметим, однако, что 490,6 кДж — это теплота, которая поглощалось бы, если бы реакция проводилась при 298 К, а не при 1800 К, как это происходит в доменной печи. Получается, что наше решение неверно? Нет, оно абсолютно верно, просто тепловой эффект (+490,6 кДж) следует рассматривать как телоту, поглощаемую при нагревании оксида железа (III) и углерода от 298 до 1800 К, последующей реакции между ними и охлаждением снова до комнатной температуры 298 К. Изменение энтальпии, или теплота реакции, зависит только от исходного и конечного состояний участников реакции, а нет от того, остается ли температура постоянной или поднимается до уровня, достигаемого в доменной печи, и затем опускается снова. Важно лишь то, что в конце процесса, как и в его начале, температура имеет значение 298 К. Еще раз повторяю, ибо это очень важный момент: Когда мы ссылаемся на теплоты реакции и утверждаем, что полученные значения относятся к процессу, проводимой «при давлении 1 атм и 298 К», требуется только, чтобы реакция начиналась при этих условиях и продукты были приведены к этим условиям (1 атм и 298 К).
Пример 2. Определите теплоту сгорания жидкого бензола.
Запишем полное уравнение реакции с указанием теплот образования всех участвующих в нем веществ:
Теплота этой реакции, как она записана (с 2 молями бензола), равна:
- ΔH = 12(-393,5) + 6(-285,8) — 2(+49,0) — 15(0,0) = -6540 кДж
Следовательно, теплота сгорания 1 моля бензола должна быть равна половине этой величины, т.е. -3270 кДж·моль -1 .
Надеюсь урок 21 «Теплота образования» был не скучным. Если у вас возникли вопросы, пишите их в комментарии.
Электронная библиотека
Ионы металлов образуют с неорганическими и органическими реагентами большое количество комплексных соединений, свойства которых широко используются в аналитической химии для идентификации, определения и маскирования.
Любое комплексное соединение состоит из центрального атома и координированных вокруг него частиц, которые называются лигандами.
Химическая связь между центральным атомом и лигандом, как правило, носит донорно-акцепторный характер, причем донором пары электронов является лиганд, а акцептором этой пары – центральный атом. Лиганд может иметь несколько донорных атомов, способных образовывать химическую связь с центральным атомом. По этому признаку все лиганды делятся на монодентатные и полидентатные.
Монодентатный лиганд занимает одно координационное место у центрального атома; полидентатный – несколько: два, три и т.д. Максимальное число монодентатных лигандов, которые могут разместиться вокруг центрального атома, носит название координационного числа атома комплексообразователя.
Центральный атом и расположенные вокруг него лиганды образуют внутреннюю координационную сферу, которую иногда называют первой координационной сферой. Внутренняя координационная сфера может иметь положительный, отрицательный или нулевой электрический заряд. Если внутренняя координационная сфера имеет заряд, мы имеем дело с комплексным катионом или анионом, и для электронейтральности комплексное соединение должно содержать анионы или катионы, которые размещаются во внешней или второй координационной сфере. Связь между внутренней и внешней координационными сферами носит чисто ионный характер. Поэтому в водных растворах ионы, находящиеся во внешней координационной сфере комплекса, полностью диссоциированы.
Лиганды представляют собой анионы или полярные молекулы. К неорганическим лигандам относят молекулы воды и аммиака, а также гидроксид-, галогенид-, цианид-ионы и т.д. Одним из наиболее распространенных лигандов является аммиак. Образование аммиачных комплексов наблюдается во всех случаях, когда происходит растворение первоначально образовавшегося осадка гидроксида или основной соли в избытке раствора аммиака. Наиболее устойчивые комплексы образуются с катионами Cu 2+ , Co 2+ , Zn 2+ , Ni 2+ , Cd 2+ , Ag + . В случае катионов, обладающих частично заполненной оболочкой, аммиакаты довольно интенсивно окрашены.
Комплексы с органическими лигандами, как правило, интенсивно окрашены, нерастворимы в воде и легко растворимы в органических средах. Обычно лиганды содержат такие донорные атомы, как кислород, азот, сера, фосфор и мышьяк, входящие в состав функциональных групп органических реагентов.
В комплексах с полидентатными лигандами могут образовываться хелатные циклы. Такие комплексы называют хелатами (от греч. chele – клешня краба). Хелаты, в которых замыкание цикла происходит в результате вытеснения ионом металла одного или нескольких протонов из кислотных групп лиганда, называют внутри комплексным и соединениями.
Успех разделения металлов во многих случаях зависит от того, насколько полно удалось перевести определяемый элемент в форму, имеющую большую константу распределения между фазами. В аналитической химии чаще всего для этих целей используется комплексообразование, кислотно-основные и окислительно-восстановительные процессы. Рассмотрим основные закономерности процессов комплексообразования.
Если ион металла поместить в воду, то в первой координационной сфере окажутся молекулы воды, которые образуют прочные химические связи с ионом металла. Мо
лекулы воды из внутренней координационной сферы обмениваются с молекулами воды из объема раствора, причем этот процесс может быть как очень быстрым, так и очень медленным.
Кинетику этого процесса можно изучать, используя меченую изотопом О 18 воду. В настоящее время имеются достоверные экспериментальные данные о том, что в первой координационной сфере ионов металлов находится строго определенное число молекул воды: 2, 4, 6 или 8. Такие ионы носят название аквоионов.
Если в раствор, содержащий аквоионы, ввести частицы, обладающие большими донорными свойствами, нежели молекулы воды, то они будут вытеснять координированные молекулы воды и, таким образом, процесс комплексообразования в водном растворе будет сводиться к реакции замещения молекул воды в аквоионе на молекулы или ионы лиганда. Процесс может быть записан следующим образом:
с константой равновесия
Здесь и далее заряды комплексной частицы и лигандов мы опускаем для упрощения записи.
Образование комплексной частицы MLn проходит ступенчато, т.е. молекулы лиганда последовательно замещают молекулы воды одну за другой, вплоть до полного вытеснения их из внутренней координационной сферы. Каждая ступень равновесия характеризуется ступенчатой или частной константой (Ki). Весь процесс характеризуется полной или общей константой (bn).
Нетрудно видеть, что bn=K1*K2* … *Kn. Уравнение материального баланса будет следующим:
Здесь СL и СМ – аналитические или полные концентрации, а в квадратных скобах равновесные концентрации соответствующих частиц.
Таким образом, мы видим, что металл может находиться в растворе в виде различных комплексных частиц, причем соотношение между ними может быть любым. Для характеристики глубины комплексообразования Бьеррумом было введено понятие среднего координационного числа ( ). Это число лигандов, связанных с ионом металла и приходящиеся в среднем на его один атом:
Совершенно очевидно, что, если лиганда нет в растворе, тогда
СL = 0 и [L] = 0 и = 0;
при добавлении лиганда часть его идет на образование комплексных частиц, а часть остается в растворе несвязанной, т.е. равновесной и растет; по мере увеличения концентрации лиганда во все большей мере образуется комплексная частица, содержащая
максимальное количество лигандов, и, наконец, когда весь металл окажется в этой форме, дальнейшее увеличение концентрации лиганда не будет приводить к увеличению количества связанного лиганда, и достигнет своего предела, равного максимальному координационному числу. Обратим внимание, что может принимать любые значения между 0 и n, как целочисленные, так и дробные.
В выражении для среднего координационного числа ( ) величины равновесных концентраций комплексных частиц можно заменить, используя константы bI, тогда получим:
Таким образом, оказывается, что для одноядерных комплексов глубина комплексообразования, характеризуемая средним координационным числом, является только функцией равновесной концентрации лиганда.
Для того, чтобы рассчитать распределение металла по различным комплексным формам, необходимо иметь данные о ступенчатых константах образования и равновесной концентрации лиганда. Равновесную концентрацию лиганда нужно либо измерить подходящим методом, либо рассчитать. Если константы образования малы и СM — на воду является неизозарядным процессом, поскольку молекула воды имеет нулевой электрический заряд.
Как всякая константа, константа образования может быть пересчитана в изобарно-изотермический потенциал:
При этом выясняется, что для многих процессов ступенчатого комплексообразования DНi – небольшая и постоянная величина. В чем же причина изменения константы при переходе от одной ступени к другой? Чем определяется величина , именуемая ступенчатым фактором. Если мы знаем эти закономерности, то довольно легко можем оценить значения ступенчатых констант, а, зная величину общей константы, можем решить обратную задачу.
Допустим, что способность соединения MLn к отщеплению лиганда пропорциональна n, а способность к присоединению лиганда пропорциональна n-i, т.е. первый процесс пропорционален числу уже присоединившихся лигандов, а второй процесс пропорционален числу вакантных мест.
Тогда для двух соседних форм отношение констант этих ступеней будет пропорционально:
Так, для n = 2 , т.е. константа первой ступени равновесия в четыре раза больше второй, если соотношение между константами определяется только этой причиной.
Таким образом, можно думать, что в любую константу входит статистический член, равный отношению вероятностей отщепления и присоединения лигандов:
тогда ступенчатую константу можно представить как .
Статистический фактор не зависит от природы частиц, а потому не имеет никакого отношения к химии, но, тем не менее, он содержится в любой ступенчатой константе. Если мы исключим статистические факторы из всех констант, то получим новые константы без учета статистики, в этом случае отношение называют лиганд-эффектом.
Часто отношение последовательных констант представляют в логарифмической форме:
Что же мы знаем о лиганд-эффекте? Оказывается, для большинства известных изозарядных процессов
Статистическое соотношение констант наблюдается во всех случаях, когда химическая природа замещаемых и замещающих лигандов не сильно отличается друг от друга. Так, при замещении Сl — на Br — независимо от металла комплексообразователя ход ступенчатых констант чисто статистический, но при замещении одного из этих лигандов на СN — появляется лиганд-эффект, отличный от нуля. Образование аминов металлов из аквокомплексов часто также подчиняется статистике. Обнаруженные при этом лиганд-эффекты не значительны, что вполне понятно, поскольку молекулы воды и аммиака имеют близкие размеры, дипольные моменты и поляризуемости.
Для неизозарядных процессов лиганд-эффект не равен нулю, и , что заставляет думать, что в лиганд-эффект входит электростатистическая составляющая, тогда как в изозарядном процессе она по определению равна нулю. Учет этой составляющей весьма сложен. Некоторыми авторами было показано, что после учета электростатистической составляющей лиганд-эффект неизозарядных процессов, как и изозарядных оказывается весьма малой величиной.
Не следует думать, что во всех случаях жизни лиганд-эффект либо равен нулю, либо представляет собой весьма малую величину. В лиганд-эффекте отражается различие в химической сущности протекающих процессов, а потому он оказывается весьма большим, когда происходят существенные химические изменения при переходе от од
ной ступени к другой, например, изменение характера химической связи, перестройка геометрической структуры частицы и т.п.
В настоящее время химия координационных соединений является одной из самых динамичных отраслей знания. Число координационных соединений реально начинает соперничать с числом органических соединений; открытие новых классов соединений, новых методов исследования, создание новых теоретических концепций происходит практически ежегодно; в этом потоке информации вряд ли есть надежда, что обнаружатся общие и в то же время достаточно конкретные закономерности, позволяющие предсказывать величины констант образования различных комплексов.
Поэтому химики выбирают более простой путь: неизвестную константу образования определяют экспериментально; далее в ряду родственных соединений по известным уже константам ищут закономерность, которая обладает определенной предсказательной силой в рамках определенного класса соединений или реакций.
Точно предсказать комплексообразующую способность элемента невозможно, даже пользуясь таким фундаментальным законом химии, как периодический закон. По-видимому, это отражает тот факт, что процессы комплексообразования связаны с очень тонкими различиями в химическом поведении элементов, и комплексообразующая способность элемента зависит как от его степени окисления, так и от природы лигандов.
В общих чертах можно утверждать, что наименьшую способность к комплексообразованию проявляют элементы нулевой группы и прилегающие к ней элементы главных подгрупп первой и седьмой группы. Наибольшая комплексообразующая способность характерна для элементов 8-й группы таблицы Менделеева. В развернутом варианте периодической таблицы наиболее типичные комплексообразователи оказываются сосредоточенными в центре таблицы.
Для главных подгрупп при одинаковом ионном радиусе устойчивость комплексов нарастает с ростом заряда; при равном заряде с ростом ионного радиуса устойчивость уменьшается. Для побочных подгрупп первое положение остается справедливым, тогда как второе меняет знак, т.е. устойчивость возрастает с ростом ионного радиуса при постоянном заряде.
Для двухзарядных металлических ионов первого переходного ряда от Mn 2+ до Zn 2+ по величине первой константы образования K1, с очень широким кругом лигандов был установлен следующий порядок устойчивости:
Этот ряд получил название ряда Эрвинга-Вильямса. Такое изменение устойчивости хорошо коррелирует с изменением второго ионизационного потенциала в этом ряду.

Наличие хорошей корреляции, по-видимому, объясняется тем, что энергия образования химической связи представляет собой, по сути, неполную компенсацию затрат на ионизацию атома. Распространение этой закономерности на элементы второго и третьего переходных рядов уже не подтверждается экспериментальными данными, что и указывает на ограниченный характер этой закономерности.
Если рассматривать процесс комплексообразования со стороны лиганда, то, прежде всего, следует отметить, что ряд лигандов не обладает специфичностью по отношению к определенным ионам металла. Сюда прежде всего относятся: вода, гидроксильный ион, ион О 2- и в некоторой мере F — . С этими лигандами все металлические ионы образуют более или менее устойчивые комплексы. Другой тип лигандов, например, молекулы, содержащие донорные атомы серы, обладают ярко выраженной специфичностью по отношении к определенным ионам металлов. Так, дитизон образует чрезвычайно устойчивые комплексы с ртутью (II) и не образует устойчивых комплексов с магнием (II).
Для ионов переходных металлов было обнаружено, что, если проводить сравнение устойчивости галогенидных комплексов по величинам bn или , то можно выделить два класса металлов. Последовательность устойчивости определяется:
для первого класса порядком ;
Первый ряд был назван обычным рядом устойчивости, а второй – обращенным. Эта закономерность была проверена на молекулах лигандов, содержащих донорные атомы: O, S, Se, Te, N, P, As, Sb. Оказалось, что во всех случаях имеется прямой и обращенный ряд устойчивости.
Те металлы, которые дают наиболее прочные комплексы с молекулами, содержащими донорные атомы F, О и N, т.е. металлы, дающие обычный ряд устойчивости, были названы металлами класса «А». Металлы, которые образуют прочные комплексы с лигандами, содержащими донорные атомы I, Te и Sb, т.е. металлы, дающие обращенный ряд устойчивости, были названы металлами класса «Б».
Металлы класса «Б» в развернутой форме периодической таблицы занимают область, напоминающую некоторым образом треугольник, причем вершина его лежит на никеле и меди, а основание треугольника простирается от осмия до висмута. Следует сказать, что, чем ниже в периодической системе находится металл, чем меньше его координационное число и чем больше степень окисления, тем выше обращенность и тем бесспорнее принадлежность этого металла к классу «Б».
При образовании хелатов акволиганды гидратированного катиона вытесняются донорными атомами хелатного лиганда. Эти представления нельзя перенести на высокозаряженные катионы небольшого размера, которые в водных растворах существуют не в виде аквоионов, а в виде оксоанионов и оксокислот.
Оксо- и гидроксолиганды вытесняются из внутренней сферы лишь немногими некислородными лигандами, например, ионом F — . Несмотря на это, высокозаряженные центральные атомы иногда образует истинные, довольно стабильные хелаты; однако их поведение отличается от поведения обычных хелатов.
В противоположность обычным хелатам, они устойчивы только в сильнокислых растворах, часто образуются даже в концентрированных кислотах и быстро разрушаются в щелочных растворах. Образование таких хелатов можно представить как образование эфира недиссоциированной гидрокислоты со спиртовыми или фенольными ОН-группами хелатного лиганда.
Особенностью хелатных комплексов является то, что они, как правило, более устойчивы по сравнению с аналогичными комплексами, образованными тем же ионом металла с монодентатными лигандами. Шварценбахом было введено понятие «хелатный эффект» для того, чтобы отразить это явление относительно более высокой устойчивости хелата металла по сравнению с аналогичными комплексами металлов с монодентатными лигандами или с хелатообразующими реагентами, но с меньшим числом хелатных циклов. Хелатный эффект определяется разностью величин lgK для сравнимых комплексов:
Для корректного сравнения необходимо, чтобы донорные атомы лигандов Z и А были одинаковы.
Для металлических ионов первого переходного ряда при сравнении устойчивости этилендиаминовых и аммиачных комплексов значения хелатного эффекта в ряду: Co 2+ , Ni 2+ , Cu 2+ и Zn 2+ составляет 2,3; 2,5; 3,0 и 1,2 логарифмической единицы соответст
венно. При переходе к пропилендиамину для Ni 2+ и Cu 2+ значения хелатного эффекта уменьшаются до 1,4 и 2,3 логарифмических единиц.
Таким образом, оказывается, что пятичленные циклы более устойчивы, чем шестичленные, а при образовании семичленных циклов хелатный эффект практически равен нулю. Пока неизвестны молекулы лигандов, содержащие донорные атомы азота и способные образовывать четырехчленные циклы в хелате. Гидразин, потенциально могущий дать трехчленный цикл, оказывается, ведет себя как монодентатный лиганд. Таким образом, можно думать, что хелатный эффект в такого рода соединениях либо равен нулю, либо отрицателен.
Первоначальное объяснение природы хелатного эффекта было следующим. Уравнения образования комплексов можно записать так:
В первом процессе из раствора уходит четыре молекулы аммиака и высвобождается четыре молекулы воды, т.е. число частиц остается неизменным. Во втором процессе на две молекулы этилендиамина высвобождаются те же четыре молекулы воды, число частиц в растворе растет, что приводит к росту энтропии системы, следовательно, существование хелатного эффекта может быть объяснено целиком энтропийным фактором.
Хотя энтропийный вклад в образование хелата всегда существует, количественное сопоставление величин хелатных эффектов приводит к тому, что мы должны признать существование и энтальпийной части его. Значение хелатного эффекта определяется следующими факторами:
1) числом хелатных циклов;
2) размером циклов;
3) изменением сольватации частиц при образовании комплекса;
4) расположением хелатных циклов;
5) изменением энтропии некоординированных лигандов;
6) изменением энтропии координированных лигандов
1) теплотой образования связи иона металла с лигандом, которая определяется электроотрицательностью иона металла и донорного атома лиганда;
2) структурой лиганда;
3) стерическим и электростатическим отталкиванием между донорными группами лигандов в комплексе;
4) кулоновским взаимодействием центрального иона с лигандами
Важнейшими факторами, которые характерны для образования хелатов и которые отличают хелатообразование от образования комплексов вообще, являются число хелатных циклов и размеры циклов.
Хотя сам процесс комплексообразования в водных растворах является реакцией замещения, образующиеся комплексные частицы могут участвовать еще в двух типах химических реакций, а именно, в кислотно-основных процессах, связанных с передачей протона, и окислительно-восстановительных, связанных с передачей электронов.
Любой аквоион металла, в соответствии с основными положениями теории кислот и оснований Бренстеда, должен быть кислотой, поскольку может быть поставщиком протонов.
Например, для аквоиона алюминия(III) процесс может быть записан следующим образом:
Обычная форма записи выглядит так:
но она отражает не существо дела, а стремление к краткости записи.
Сама молекула воды обладает кислотными свойствами и имеет константу диссоциации порядка 10 -16 . При координации вблизи заряженного иона металла, естественно, способность ее к диссоциации должна возрасти. Так, в случае аквоиона А1(III) мы имеем рК1=4,9; Fе(III) – 2,5; Cr(III) – 4,0; Mg(II) – 12,1; Ca(II) – 12,8; Cu(II) – 7,0; Ag(I) – 11,7; Hg(II) – 2,8. Из этих данных видно, что способность к диссоциации координированных молекул воды увеличивается на 3 – 13 порядков, причем, чем выше заряд иона, тем сильнее кислота.
Процесс кислотной диссоциации аквоиона формально можно рассматривать как процесс замещения координированных молекул воды на гидроксильный ион:
с константой равновесия
Если теперь учесть константу автопротолиза воды:
Таким образом, термодинамически безразлично, как описывать кислотные свойства аквоионов: в виде процесса диссоциации или замещения на гидроксильный ион. Решить вопрос о том, как же в действительности идет процесс, помогают кинетические исследования. Дело в том, что процессы диссоциации идут с очень большими скоростями, тогда как скорости реакций замещения на несколько порядков меньше. Это позволяет утверждать, что в большинстве случаев реализуется механизм диссоциации.
Кислотные свойства координированной молекулы зависят не только от общего заряда комплекса, но и от ближайшего окружения во внутренней координационной сфере. Так, например, [Со(Н2О)6] 3+ – очень сильная кислота, тогда как [Со(NН3)5Н2O] 3+ имеет рК1
5. Аквоион Rh(III) [Rh(Н2O)6] 3+ – кислота средней силы с рК1
3, в то время, как ион [RhCl5Н2O] 2- – очень слабая кислота с рК1
При оценке кислотных свойств комплексных соединений следует всегда иметь в виду, что способность лиганда отщеплять протон при координации всегда возрастает, если он находится вблизи донорного атома. Например, вода увеличивает свои кислотные свойства, сероводород и галогеноводородные кислоты ведут себя таким же образом, поэтому комплексные кислоты оказываются более сильными кислотами, нежели обычные.
Положение меняется, когда между донорным атомом и протонсодержащей группой находится хотя бы еще один атом, который не может передавать влияние центрального иона. Например, аминокислоты могут координироваться не только биден
татно, но и монодентатно, причем координация осуществляется через атом азота аминогруппы. Кислотные свойства некоординированной карбоксильной группы оказываются такими же, как и в некоординированной кислоте. Причина этого заключается в том, что группа СН2 не передает влияние центрального иона на карбоксильную группу.
Заканчивая рассмотрение кислотных свойств комплексов, можно поставить вопрос: каким образом рН среды влияет на процессы комплексообразования? Здесь можно выделить два случая:
· первый, когда лиганд является анионом сильной кислоты;
· второй, когда он представляет собой анион слабой кислоты.
В первом случае нет специфического влияния концентрации водородного иона, она влияет так же, как и изменение концентрации любого другого иона через изменение коэффициентов активности. Во втором случае процесс представляет собой сопряженные процессы диссоциации слабой кислоты и образования комплекса:
Глубина образования МL однозначно определяется равновесной концентрацией [L — ], которую следует определить из уравнения диссоциации слабой кислоты.
Роль рН в этом процессе сводится к созданию определенной равновесной концентрации лиганда и, тем самым, определяет глубину комплексообразования. В кислых средах [L — ]®0, и комплекс не образуется, в щелочных средах [L — ]®СL, и глубина комплексообразования будет определяться только значением K1 при заданном значении СL.
Комплексные соединения участвуют в окислительно-восстановительных реакциях, отличительной чертой которых является передача электронов от восстановителя к окислителю. Каким же образом комплексообразование влияет на окислительный потенциал частицы? Как известно, окислительный потенциал системы определяется из уравнения Нернста:
Для системы Аg + /Ag° это уравнение будет выглядеть:
поскольку [Ag°] – это собственная растворимость металлического серебра, то – величина постоянная и может быть объединен с . Тогда имеем:
Если в систему Аg + /Ag° ввести лиганд, например аммиак, то ионы серебра будут связываться в комплекс, равновесная концентрация серебра будет падать и окислительный потенциал будет понижаться:
Из уравнения (4.1) видно, что окислительный потенциал уменьшается с ростом равновесной концентрации лиганда, а нормальный стандартный потенциал комплексной формы отличается от стандартного потенциала аквоиона на величину .
Отсюда открывается возможность регулировать окислительную способность иона путем простой замены ближайшего окружения, т.е. путем проведения реакций комплексообразования. Так, за счет реакций комплексообразования мы можем превратить один из самых сильных окислителей аквоион золота(I) [Au(H2O)2] + (E0 = 1,68 В) в сильный восстановитель – цианидный комплекс золота(I) [Au(CN)2] ‑ (E0 = -0,60 В).
Если концентрация восстановленной формы в уравнении Нернста не является постоянной величиной, как, например, для системы Fe 3+ /Fe 2+ , предсказать изменение окислительных свойств труднее. Уравнение Нернста для системы имеет следующий вид:
В том случае, когда лиганд образует более прочные комплексы с ионом Fe 3+ , потенциал будет падать, если же прочнее комплексы с ионом Fe 2+ , потенциал будет расти, по сравнению с потенциалом аквоионов.
Заканчивая обсуждение вопросов комплексообразования, можно сказать, что эти процессы дают в руки аналитика очень эффективные средства тонкой регулировки химической природы частиц. С их помощью катионы можно превращать в анионы или нейтральные частицы, сильно гидратированные ионы – в ионы слабо гидратированные, гидролизующиеся ионы – в ионы негидролизующиеся и, наконец, окислители – в восстановители. Все это привело к тому, что в настоящее время подавляющее большинство химиков-аналитиков занимается проблемами координационной химии.
Метод молекулярных орбиталей. Основные положения метода
» data-shape=»round» data-use-links data-color-scheme=»normal» data-direction=»horizontal» data-services=»messenger,vkontakte,facebook,odnoklassniki,telegram,twitter,viber,whatsapp,moimir,lj,blogger»>
Метод молекулярных орбиталей (ММО, МО).
Хронологически метод молекулярных орбиталей появился позже метода валентных связей, поскольку оставались в теории ковалентной связи вопросы, которые не могли получить объяснение методом ВС. Укажем некоторые из них.
Как известно, основное положение метода ВС состоит в том, что связь между атомами осуществляется за счет электронных пар (связующих двухэлектронных облаков). Но это не всегда так. В ряде случаев в образовании химической связи участвуют отдельные электроны. Так, в молекулярном ионе Н2 + одноэлектронная связь. Метод ВС образование одноэлектронной связи объяснить не может, она противоречит его основному положению.
Метод ВС не объясняет также роли неспаренных электронов в молекуле. Молекулы, имеющие неспаренные электроны, парамагнитны, т. е. втягиваются в магнитное поле, так как неспаренный электрон создает постоянный магнитный момент. Если в молекулах нет неспаренных электронов, то они диамагнитны – выталкиваются из магнитного поля. Молекула кислорода парамагнитна, в ней имеется два электрона с параллельной ориентацией спинов, что противоречит методу ВС. Необходимо также отметить, что метод ВС не смог объяснить ряд свойств комплексных соединений – их цветность и др.
Чтобы объяснить эти факты, был предложен метод молекулярных орбиталей (ММО).
4.5.1. Основные положения ММО, МО.
1. В молекуле все электроны являются общими. Сама молекула — это единое целое, совокупность ядер и электронов.
2. В молекуле каждому электрону соответствует молекулярная орбиталь, подобно тому как каждому электрону в атоме соответствует атомная орбиталь. И обозначения орбиталей аналогичны:

3. В первом приближении молекулярная орбиталь представляет собой линейную комбинацию (сложение и вычитание) атомных орбиталей. Поэтому говорят о методе молекулярных орбиталей ЛКАО (молекулярная орбиталь есть линейная комбинация атомных орбиталей), при которой из N АО образуется N МО (это основное положение метода).

Рис. 12. Энергетическая схема образования молекулы водорода Н2
Рассмотрение химических связей в методе молекулярных орбиталей заключается в распределении электронов в молекуле по ее орбиталям. Последние заполняются в порядке возрастания энергии и с учетом принципа Паули. В этом методе предполагается увеличение электронной плотности между ядрами при образовании ковалентной связи.
Пользуясь положениями 1—3, объясним образование молекулы H2 с точки зрения метода молекулярных орбиталей. При достаточном сближении атомов водорода происходит перекрывание их электронных орбиталей. Согласно п. 3 из двух одинаковых ls-орбиталей образуются две молекулярные орбитали: одна из них от сложения атомных орбиталей, другая от их вычитания (рис.12). Энергия первой E1 св ), а находящиеся на ней электроны—связывающими электронами.
Молекулярная орбиталь, энергия которой больше энергии атомной орбитали, называется антисвязывающей или разрыхляющей (обозначается символом разр ), а находящиеся на ней электроны — разрыхляющими электронами.
Если у соединяющихся атомов водорода спины электронов антипараллельны, то они займут связывающую молекулярную орбиталь, возникает химическая связь (рис. 12), сопровождающаяся выделением энергии E1 (435 кДж/моль). Если же спины электронов атомов водорода параллельны, то они в соответствии с принципом Паули не могут разместиться на одной молекулярной орбитали: один из них разместится на связывающей, а другой на разрыхляющей орбитали, значит химическая связь образоваться не может.
Согласно методу молекулярных орбиталей образование молекул возможно, если число электронов на связывающих орбиталях больше числа электронов на разрыхляющих орбиталях. Если же число электронов на связывающих и разрыхляющих орбиталях одинаково, то такие молекулы образоваться не могут. Так, теория не допускает существования молекулы Нe2, так как в ней два электрона находились бы на связывающей орбитали и два — на разрыхляющей. Всегда разрыхляющий электрон сводит на нет действие связывающего электрона.
В системе обозначений метода молекулярных орбиталей реакцию образования молекулы водорода из атомов записывают так:
2H[1s 1 ] = H2[(σ CB 1s) 2 ],
т. е. используются символы, выражающие размещение электронов на атомных и молекулярных орбиталях. При этом символ каждой молекулярной орбитали заключается в круглые скобки и над скобками справа проставляется число электронов на этой орбитали.
Число валентных связей определяется по формуле:

В молекуле водорода В = (2—0) : 2=1, водород одновалентен. Молекула Н2 диамагнитна (электроны спарены).
Теперь легко объясняется одноэлектронная связь в молекулярном ионе Н2 + (рис.13). Единственный электрон этого иона занимает энергетически наиболее выгодную орбиталь св 1s. Уравнение процесса:
H[1s 1 ] + H + = H2 + [(σ св 1s) 1 ], ∆H = – 259,4 кДж
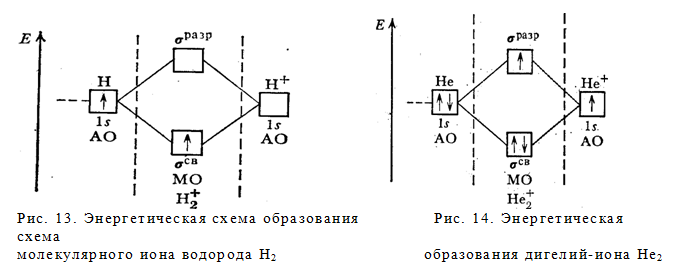
Число связей в ионе H2 + равно ½ (связь одним электроном). Ион H2 + — парамагнитен (имеет один неспаренный электрон).
Возможно существование молекулярного дигелий иона Не2 + (рис.14). Уравнение его образования
He[1s 2 ] + He + [1s 1 ] = He2 + [(σ CB 1s) 2 (σ разр 1s) 1 ], ∆H = – 292,8 кДж
Этот ион экспериментально обнаружен. Число связей в нем (2—1) : 2 = 1 /2. Ион— парамагнитен (имеет неспаренный электрон).

Рис. 15 . Энергетическая схема образования двухатомных гомонуклеарных молекул элементов второго периода
4.5.2. Основные двухатомные гомонуклеарные молекулы элементов 2-го периода. Рассмотренный принцип построения молекулярных орбиталей из двух одинаковых АО сохраняется при построении гомонуклеарных молекул элементов 2-го периода системы Д.И. Менделеева. Они образуются в результате взаимодействия 2s- и 2рx-, 2рy– и 2рz-орбиталей.
Участием внутренних электронов 1s-орбиталей можно пренебречь (на последующих энергетических схемах они не учтены). 2s-орбиталь одного атома взаимодействует только с 2s-орбиталью другого атома (должна быть близость значений энергий взаимодействующих орбиталей), образуя МО σ2s св и σ2s разр . При перекрывании (взаимодействии) 2р-орбиталей обоих атомов образуются МО: σх св , σх разр , πу св , πу разр , πz св , πz разр
(рис.15). Т.е. из шести исходных 2р-орбиталей образуется шесть молекулярных орбиталей – три связывающих и три разрыхляющих. Молекулярные орбитали, образующиеся из s- и рx-атомных орбиталей, обозначаются буквой , а из ру– и рz– – буквой . С помощью рис. 15 легко представить электронные конфигурации этих молекул в системе обозначений метода молекулярных орбиталей.

Рис. 16. Энергетическая схема образования молекулы Li2
Пример 1. Молекула лития Li2. Схема ее образования представлена на рис.16. В ней два связывающих электрона, молекула диамагнитна (электроны спарены). Написание уравнения и формулы можно упростить, обозначив внутренний уровень через K:
2Li[K2s] = Li2[KK(σ CB 2s) 2 ]
Число связей равно 1.
Пример 2. Молекула бериллия Be2. Восемь электронов молекулы размещены на МО следующим образом:
Ве2[KK(σ CB 2s) 2 (σ разр 2s) 2 ]
Как видно, число связей в молекуле равно нулю: два разрыхляющих электрона уничтожают действие двух связывающих. Такая молекула не может существовать, и она до сих пор не обнаружена. Необходимо отметить, что невозможны двухатомные молекулы у всех элементов IIА-группы, палладия и инертных элементов, так как их атомы имеют замкнутую электронную структуру.
Пример 3. Молекула азота N2 (рис. 17). Распределение 14 электронов по МО записывается так:


Рис. 17. Энергетическая схема образования молекулы N2
Под формулой указано число связей в молекуле, исходя из расчета, что два электрона, расположенные на одной МО, образуют валентную связь; знак плюс обозначает связующие орбитали, знак минус – разрыхляющие. Число связей в молекуле 3. нет неспаренных электронов – молекула диамагнитна.
Пример 4. Молекула O2 (рис. 18). Электроны размещаются по молекулярным орбиталям в последовательности:
В молекуле две валентные связи. Последние два электрона разместились на различных π-разрыхляющих орбиталях в соответствии с правилом Гунда. Два неспаренных электрона обусловливают парамагнетизм молекулы кислорода.


Рис. 18. Энергетическая схема образования молекулы O2
4.5.3. Двухатомные гетеронуклеарные молекулы элементов 2-го периода. Энергетическая схема образования МО гетеронуклеарных двухатомных молекул, состоящих из атомов элементов 2-го периода, представлена на рис. 19. Она сходна со схемой образования молекулярных орбиталей гомонуклеарных молекул.
Основное различие сводится к тому, что значения энергии одноименных орбиталей атомов разных элементов не равны между собой, поскольку различны заряды ядер атомов. В качестве примера рассмотрим электронную валентную конфигурацию молекул СО и NO.

Рис. 19 . Энергетическая схема образования двух атомных гетеронуклеарных молекул элементов второго периода
Пример 5.Молекула СО. Внешняя электронная оболочка атома углерода имеет конфигурацию 2s 2 2p 2 , а кислорода 2s 2 2p 4 . Стало быть, в заполнении МО молекулы СО принимают участие 4+6=10 электронов. Из них два размещаются на орбитали σ2s св , два – на орбитали σ2s разр , четыре – на орбиталях πy CB и πz CB , а девятый и десятый – на σх св . Таким образом, электронную валентную конфигурацию молекулы СО можно выразить формулой:

Как и предусматривалось теорией ВС, в молекуле СО три валентные связи (сравните с N2). Молекула диамагнитна – все электроны спарены.
Пример 6. Молекула NO. На МО молекулы оксида азота (II) должны разместиться 11 электронов: пять азота – 2s 2 2p 3 и шесть кислорода – 2s 2 2p 4 . Десять из них размещаются так же, как и электроны молекулы оксида углерода (II) (пример 5), а одиннадцатый разместится на одной из разрыхляющих орбиталей – πy разр или πZ разр (эти орбитали энергетически эквивалентны между собой). Тогда
Значит, молекула NO имеет две с половиной валентные связи, энергия связи большая —677,8кДж/моль. Она парамагнитна, так как содержит один неспаренный электрон.

Приведенные примеры служат иллюстрацией возможностей метода МО в объяснении строения и свойств молекул.
Пример 7. Какую валентность, обусловленную неспаренными электронами (спинвалентность), может проявлять фосфор в нормальном и возбужденном состояниях?
Решение. Распределение электронов внешнего энергетического уровня фосфора 3s 2 3р 3 (учитывая правило Хунда, ) по квантовым ячейкам имеет вид:

Атомы фосфора имеют свободные d-орбитали, поэтому возможен переход одного 3s-электрона в 3d-состояние:

Отсюда валентность (спинвалентность) фосфора в нормальном состоянии равна трем, а в возбужденном — пяти.
Пример 8. Что такое гибридизация валентных орбиталей? Какое строение имеют молекулы типа АВn, если связь в них образуется за счет sp-, sp 2 -, sp 3 -гибридизации орбиталей атома А?
Решение. Теория валентных связей (ВС) предполагает участие в образовании ковалентных связей не только чистых АО, но и смешанных, так называемых гибридных, АО. При гибридизации первоначальная форма и энергия орбиталей (электронных облаков) взаимно изменяются и образуются орбитали (облака) новой одинаковой формы и с одинаковой энергией. Число гибридных орбиталей (q) равно числу исходных. Ответ см. в табл. 13.
Гибридизация орбиталей и пространственная конфигурация молекул

Пример 9. Как метод молекулярных орбиталей (МО) описывает строение двухатомных гомоядерных молекул элементов второго периода?
Решение. Метод валентных связей (ВС) не может объяснить целый ряд свойств и строение некоторых молекул (парамагнетизм молекулы О2; большую прочность связей в молекулярных ионах F + 2 и О + 2, чем, соответственно, в молекулах F2 и O2; наоборот, меньшую прочность связи в ионе N + 2 , чем в молекуле N2; существование молекулярного иона Не2 + и неустойчивость молекулы Не2 и т.п.). Более плодотворным оказался другой подход к объяснению ковалентной связи — метод молекулярных орбиталей (МО). В методе МО состояние молекулы описывается как совокупность электронных молекулярных орбиталей. При этом число молекулярных орбиталей равно сумме атомных орбиталей. Молекулярной орбитали, возникающей от сложения атомных орбиталей (АО), соответствует более низкая энергия, чем исходным орбиталям. Такая МО имеет повышенную электронную плотность в пространстве между ядрами, способствует образованию химической связи и называется связывающей. Молекулярной орбитали, образовавшейся от вычитания атомной, соответствует более высокая энергия, чем атомной орбитали. Электронная плотность в этом случае сконцентрирована за ядрами атомов, а между ними равна нулю. Подобные МО энергетически менее выгодны, чем исходные АО, они приводят к ослаблению химической связи и называются разрыхляющими. Электроны, занимающие связывающие и разрыхляющие орбитали, называют соответственно связывающими (cв) и разрыхляющими (разр). Заполнение молекулярных орбиталей происходит при соблюдении принципа Паули и правила Хунда по мере увеличения их энергии в такой последовательности:
σ CB 1s разр 1s CB 2s разр 2s CB 2px CB 2py =
На рис. 20 изображена энергетическая схема образования молекулярных орбиталей из атомных для двухатомных гомоядерных (одного и того же элемента) молекул элементов второго периода. Число связывающих и разрыхляющих электронов зависит от их числа в атомах исходных элементов.
Следует отметить, что при образовании молекул N2 энергия связывающей 2рx-орбитали больше энергии связывающих 2ру– и 2рz-орбиталей, тогда как в молекулах О2 и F2, наоборот, энергия связывающих 2ру– и 2рz-орбиталей больше энергии связывающей 2рx-орбитали. Это нужно учитывать при изображении энергетических схем (см. рис. соответствующих молекул).

Рис.20. Энергетическая схема образования молекулярных орбиталей из атомных для гомоядерных молекул второго периода
Порядок связи в молекуле определяется разностью между числом связывающих и разрыхляющих электронов, деленной на два. Порядок связи может быть равен нулю (молекула не существует), целому или дробному положительному числу.
Подобно электронным формулам, показывающим распределение электронов в атоме по атомным орбиталям, в методе МО составляют формулы молекул, отражающие их электронную конфигурацию. По аналогии с атомными s-, p-, d-, f-орбиталями молекулярные орбитали обозначаются греческими буквами . Так, электронная конфигурация молекулы O2 записывается следующим образом:
Буквами КК показано, что четыре 1s-электрона (два связывающих и два разрыхляющих) практически не оказывают влияния на химическую связь.
http://libraryno.ru/4-processy-kompleksoobrazovaniya-anhim/
http://farmf.ru/lekcii/metod-molekulyarnyh-orbitalej-osnovnye-polozheniya-metoda/