БЕЗОПАСНОСТЬ ДОРОЖНОГО ДВИЖЕНИЯ В ЭКЗАМЕНАЦИОННЫХ БИЛЕТАХ И В ЖИЗНИ
Билет 1
Билет 10
БИЛЕТ 10. Граничные условия для векторов Е и D . Преломление силовых линий на границе диэлектриков.
Билет 10. Сварка трением
Билет 11
БИЛЕТ 11 1.Теоретические труды В.К.Тредиаковского в связи с литературными представлениями классицизма.
Билет 11. Способы сварки плавлением
Билет 12
Билет 12
1. Реакции нуклеофильного присоединения. Гетеролитические реакции с участием π-связи углерод-кислород (альдегиды, кетоны). Реакции карбонильных соединений с водой, спиртами, тиолами, первичными аминами. Роль кислотного катализа. Гидролиз ацеталей и иминов. Реакции альдольного присоединения, расщепления. Биологическое значение этих процессов.
Для альдегидов и кетонов наиболее характерны реакции нуклеофильного присоединения AN.
Общее описание механизма нуклеофильного присоединения AN
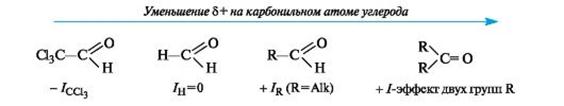
Легкость нуклеофильной атаки по атому углерода карбонильной группы альдегида или кетона зависит от величины частичного
положительного заряда на атоме углерода, его пространственной доступности и кислотно-основных свойств среды.
С учетом электронных эффектов групп, связанных с карбонильным атомом углерода, величина частичного положительного заряда δ+ на нем в альдегидах и кетонах убывает в следующем ряду:
Пространственная доступность карбонильного атома углерода уменьшается при замене водорода более объемистыми органиче- скими радикалами, поэтому альдегиды более реакционноспособны, чем кетоны.
Общая схема реакций нуклеофильного присоединения AN к карбонильной группе включает нуклеофильную атаку по карбонильному атому углерода, за которой следует присоединение электрофила к атому кислорода.
В кислой среде активность карбонильной группы, как правило, увеличивается, поскольку вследствие протонирования атома кислорода на атоме углерода возникает положительный заряд. Кислотный катализ используют обычно тогда, когда атакующий нуклеофил обладает низкой активностью.
По приведенному выше механизму осуществляется ряд важных реакций альдегидов и кетонов.
Многие свойственные альдегидам и кетонам реакции протекают в условиях организма, эти реакции представлены в последующих разделах учебника. В настоящей главе будут рассмотрены наиболее важные реакции альдегидов и кетонов, которые в обзорном виде приведены на схеме.
Присоединение спиртов.Спирты при взаимодействии с альдегидами легко образуют полуацетали.Полуацетали обычно не выделяют из-за их неустойчивости. При избытке спирта в кислой среде полуацетали превращаются в ацетали.
Применение кислотного катализатора при превращении полуацеталя в ацеталь становится понятным из приведенного ниже механизма реакции. Центральное место в нем занимает образование карбо- катиона (I), стабилизированного за счет участия неподеленной пары электронов соседнего атома кислорода (+M-эффект группы С2Н5О).
Реакции образования полуацеталей и ацеталей обратимы, поэтому ацетали и полуацетали легко гидролизуются избытком воды в кислой среде. В щелочной среде полуацетали устойчивы, так как алкоксидион является более трудно уходящей группой, чем гидроксид-ион.
Образование ацеталей часто используется как временная защита альдегидной группы.
Присоединение воды.Присоединение воды к карбонильной группе – гидратация – обратимая реакция. Степень гидратации альдегида или кетона в водном растворе зависит от строения субстрата.
Продукт гидратации, как правило, в свободном виде выделить с помощью перегонки не удается, так как он разлагается на исходные компоненты. Формальдегид в водном растворе гидратирован более чем на 99,9%, ацетальдегид – приблизительно наполовину, ацетон практически не гидратирован.
Формальдегид (муравьиный альдегид) обладает способностью свертывать белки. Его 40% водный раствор, называемый формалином, применяется в медицине как дезинфицирующее средство и консервант анатомических препаратов.
Трихлороуксусный альдегид (хлораль) гидратирован полностью. Электроноакцепторная трихлорометильная группа настолько стабилизирует хлоральгидрат, что это кристаллическое вещество отщепляет воду только при перегонке в присутствии дегидратирующих веществ – серной кислоты и др.
В основе фармакологического эффекта хлоральгидрата СС13СН(ОН)2 лежит специфическое действие на организм альдегидной группы, обусловливающее дезинфицирующие свойства. Атомы галогена усиливают ее действие, а гидратация карбонильной группы снижает токсичность вещества в целом.
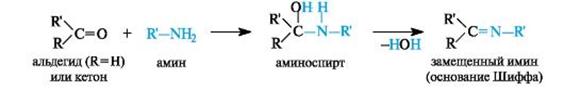
Присоединение аминов и их производных.Амины и другие азотсодержащие соединения общей формулы NH2X (X = R, NHR) реагируют с альдегидами и кетонами в две стадии. Сначала образуются продукты нуклеофильного присоединения, которые затем вследствие неустойчивости отщепляют воду. В связи с этим данный процесс в целом классифицируют как реакцию присоединения-отщепления.
В случае первичных аминов получаются замещенные имины (их называют также основаниями Шиффа).
Имины – промежуточные продукты многих ферментативных процессов. Получение иминов проходит через стадию образования аминоспиртов, которые бывают относительно устойчивы, например при взаимодействии формальдегида с α-аминокислотами.
Имины являются промежуточными продуктами получения аминов из альдегидов и кетонов путём восстановительного аминирования. Этот общий способ заключается в восстановлении смеси карбонильного соединения с аммиаком (или амином). Процесс протекает по схеме присоединения-отщепления с образованием имина, который затем восстанавливается в амин.
При взаимодействии альдегидов и кетонов с производными гидразина получаются гидразоны. Эту реакцию можно использовать для выделения альдегидов и кетонов из смесей и их хроматографической идентификации.
Основания Шиффа и другие подобные соединения легко гидролизуются водными растворами минеральных кислот с образованием исходных продуктов.
В большинстве случаев для реакций альдегидов и кетонов с азотистыми основаниями необходим кислотный катализ, ускоряющий дегидратацию продукта присоединения. Однако если слишком повысить кислотность среды, то реакция замедлится в результате превращения азотистого основания в нереакционноспособную сопряженную кислоту XNH3+.
Наличие СН-кислотного центра в молекуле альдегида или кетона приводит к тому, что α-атомы водорода этих карбонильных соединений обладают некоторой протонной подвижностью. Под действием оснований такие протоны могут отщепляться с образованием соответствующих карбанионов. Карбанионы играют роль нуклеофилов по отношению к карбонильному субстрату. Это обусловливает возможность осуществления реакций, в которых одна молекула в качестве нуклеофила присоединяется к карбонильной группе другой молекулы нейтрального карбонильного соединения. Такие процессы относятся к реакциям конденсации.
Конденсацией называют реакцию, приводящую к возникновению новой углерод-углеродной связи, причем из двух или нескольких относительно простых молекул образуется новая, более сложная молекула.
Так, в щелочной среде из двух молекул ацетальдегида образуется гидроксиальдегид с удвоенным числом атомов углерода.
Продукт реакции, содержащий гидроксильную и альдегидную группы, называется альдолем (от слов альдегид и алкоголь), а сама реакция получила название альдольной конденсации, или альдольного присоединения.
При действии основания в карбонильном соединении отщепляется протон из α-положения и образуется карбанион (I), в котором отрицательный заряд делокализован при участии карбонильной группы.
Анион (I) представляет собой сильный нуклеофил (на следующей стадии механизма он показан цветом), который присоединяется ко второй (неионизированной) молекуле карбонильного соединения. В результате такого взаимодействия возникает новая связь С-С и образуется промежуточный алкоксид-ион (II). В водной среде этот анион стабилизируется, отщепляя протон от молекулы воды, и превращается в конечный продукт – альдоль.
Реакция альдольного присоединения показана на примере пропаналя (цветом выделена молекула, присоединяющаяся к группе С=О другой молекулы); аналогичная реакция приведена на примере ацетона.
Продукт конденсации – альдоль – способен к отщеплению воды с образованием α,β-ненасыщенного карбонильного соединения. Обычно это происходит при повышенной температуре. В этом случае реакция в целом называется кротоновой конденсацией.
Реакции конденсации могут протекать и в смешанном варианте, с использованием разных карбонильных соединений, причем одно из них может и не содержать СН-кислотного центра, как, например, формальдегид и бензальдегид в следующих реакциях:
Альдольная конденсация – обратимая реакция; обратный процесс называется альдольным расщеплением (или ретроальдольной реакцией). Обе реакции происходят во многих биохимических процессах.
2. Нуклеозиды. Гидролиз нуклеозидов. Нуклеотиды. Строение мононуклеотидов, образующих нуклеиновые кислоты. Гидролиз нуклеотидов. Рибонуклеиновые и дезоксирибонуклеиновые кислоты (РНК, ДНК). Роль фодородных связей в формировании вторичной структуры ДНК.
В химии нуклеиновых кислот входящие в их состав гетероциклические соединения пиримидинового и пуринового рядов обычно называют нуклеиновыми основаниями. Нуклеиновые основания в качестве заместителей в гетероцикле могут содержать: либо оксогруппу, как в урациле и тимине; либо аминогруппу, как в аденине; либо одновременно обе эти группы, как в цитозине и гуанине.
Кислородсодержащие основания представлены лактамными таутомерными формами, в которых ароматичность не нарушена. Для всех оснований приняты сокращенные трехбуквенные обозначения, составленные из первых букв их латинских названий.
Нуклеиновые кислоты различаются входящими в них гетероциклическими основаниями: урацил входит только в РНК, а тимин – в ДНК:
Нуклеиновые основания образуют связь за счет одного из атомов азота с аномерным центром пентозы (D-рибозы или 2-дезокси-D- рибозы). Этот тип связи аналогичен обычной гликозидной связи и известен как N-гликозидная связь, а сами гликозиды – как N-гликозиды. В химии нуклеиновых кислот их называют нуклеозидами. В состав природных нуклеозидов пентозы входят в фуранозной форме (атомы углерода в них нумеруют цифрой со штрихом). Гликозидная связь осуществляется с атомом азота N-1 пиримидинового и N-9 пуринового оснований.
Природные нуклеозиды всегда являются β-аномерами. В зависимости от природы углеводного остатка различают рибонуклеозиды и дезоксирибонуклеозиды. Для нуклеозидов употребительны названия, производимые от тривиального названия соответствующего нуклеинового основания с суффиксами -идин у пиримидиновых и -озин у пуриновых нуклеозидов.
Исключение составляет название «тимидин» (а не дезокситимидин), используемое для дезоксирибозида тимина, входящего в состав ДНК. В тех редких случаях, когда тимин встречается в РНК, соответствующий нуклеозид называется риботимидином. Трехбуквенные символы нуклеозидов отличаются от символов оснований последней буквой. Однобуквенные символы применяются только для остатков (радикалов) нуклеозидов в более сложных структурах. Нуклеозиды устойчивы к гидролизу в слабощелочной среде, но гидролизуются в кислой. Пуриновые нуклеозиды гидролизуются легко, пиримидиновые труднее.
Нуклеотидами называют фосфаты нуклеозидов. Фосфорная кислота обычно этерифицирует спиртовый гидроксил при С-5′ или С-3′ в остатке рибозы (рибонуклеотиды) или дезоксирибозы (дезоксирибонуклеотиды). Общий принцип строения нуклеотидов показан на примере фосфатов аденозина. Для связывания трех компонентов в молекуле нуклеотида используются сложноэфирная и N-гликозидная связи. Нуклеотиды можно рассматривать, с одной стороны, как эфиры нуклеозидов (фосфаты), а с другой – как кислоты (в связи с наличием остатка фосфорной кислоты).
За счет фосфатного остатка нуклеотиды проявляют свойства двухосновной кислоты и в физиологических условиях при рН
7 находятся в полностью ионизированном состоянии.
Для нуклеотидов используют два вида названий (табл. 14.1). Одно включает наименование нуклеозида с указанием положения в нем фосфатного остатка, например, аденозин-3′-фосфат, уридин-5′-фос- фат; другое строится с добавлением сочетания -иловая кислота к названию остатка пиримидинового основания, например, 5′-уридило- вая кислота, или пуринового основания, например 3′-адениловая кислота. Используя принятый для нуклеозидов однобуквенный код, 5′-фосфаты записывают с добавлением латинской буквы «р» перед символом нуклеозида, 3′-фосфаты – после символа нуклеозида. Аденозин-5′-фосфат обозначается рА, аденозин-3′-фосфат – Ар и т. п. Эти сокращенные обозначения используют для записи последовательности нуклеотидных остатков в нуклеиновых кислотах. По отношению к свободным нуклеотидам в биохимической литературе широко используют их названия как монофосфатов с отражением этого признака в сокращенном коде, например АМР (или АМФ) для аденозин-5′-фосфата и т. д. (см. табл. 14.1).
К циклофосфатам относятся нуклеотиды, у которых одна молекула фосфорной кислоты этерифицирует одновременно две гидроксильные группы углеводного остатка. Практически во всех клетках присутствуют два нуклеозидциклофосфата – аденозин-3′,5′- циклофосфат (cAMP) и гуанозин-3′,5′-циклофосфат (cGMP). В полинуклеотидных цепях нуклеотидные звенья связаны через фосфатную группу. Фосфатная группа образует две сложноэфирные связи: с С-3′ предыдущего и с С-5′ последующего нуклеотидных звеньев. Каркас цепи состоит из чередующихся пентозных и фосфатных остатков, а гетероциклические основания являются «боковыми» группами, присоединенными к пентозным остаткам. Нуклеотид со свободной 5′-ОН группой называют 5′-концевым, а нуклеотид со свободной З’-ОН группой – З’-концевым. Принцип построения цепи РНК такой же, как и у ДНК, с двумя исключениями: пентозным остатком в РНК служит D-рибоза, а в наборе гетероциклических оснований используется не тимин, а урацил. Первичная структура нуклеиновых кислот определяется последовательностью нуклеотидных звеньев, связанных ковалентными связями в непрерывную цепь полинуклеотида.
Важной характеристикой нуклеиновых кислот служит нуклеотидный состав, т.е. набор и количественное отношение нуклеотидных компонентов. Нуклеотидный состав устанавливают, как правило, путем исследования продуктов гидролитического расщепления нуклеиновых кислот. Водородные связи участвуют в формировании вторичной и третичной структуры белка, а также связывают друг с другом две спирали ДНК.
Нуклеозиды значительно лучше растворимы в воде, чем исходные азотистые основания. Подобно всем гликозидам, нуклеозиды устойчивы к действию щелочей, но при нагревании легко подвергаются кислотному гидролизу с разрывом гликозидной связи и образованием основания и пентозы:
Пиримидиновые нуклеозиды значительно более устойчивы к гидролизу, чем пуриновые. В условиях in vivo гидролиз обоих типов нуклеозидов осуществляется при помощи специфических ферментов, называемых нуклеозидазами.
3. Приведите проекционную формулу стереоизомера природной α-аминомасляной кислоты, содержащей алифатический радикал, в форме биполярного иона.
Природный стереоизомер (L-форма) D-форма
H3N + Н Н NH3 +
4. Напишите уравнение реакции гистидина с азотистой кислотой.
+ НО – N = O → CH2–CH – COOH
5. Получите β-гидроксимасляную кислоту из этаналя. Укажите условия реакций. Какие вам известны биологически важные реакции, протекающие по типу альдольной конденсации?
Интересно, что в живых организмах альдольная реакция также используется достаточно часто. Например, она входит в последовательность стадий биосинтеза глюкозы — глюконеогенез, а также в обратный процесс гликолиза, который приводит к разложению глюкозы. Подобные процессы в организмах катализируются специальными ферментами — альдолазами.
Дата добавления: 2015-02-10 ; просмотров: 321 ; Нарушение авторских прав
Уравнение реакции гистидина с азотистой кислотой
Гистидин – условно незаменимая гетероциклическая α-аминокислота, одна из 20 протеиногенных аминокислот.
Гистидин — 2-амино-3-имидазолилпропановая или α-амино-β-имидазолилпропионовая кислота.
Гистидин (Гис,His,H) — аминокислота со слабыми основными свойствами, обусловленными присутствием в молекуле остатка имидазола, молекулярная формула — C₆H₉N₃O₂.
Гистидин был выделен в 1896 году одновременно двумя учѐными: Kossel из сернокислых гидролизатов протамина спермы осетра и Hedin – из белковых гидролизатов.
Суточная потребность
Дневная потребность в гистидине составляет 1,5 – 2,0 грамма.
Физические свойства
Гистидин представляет собой прозрачные, бесцветные или белые кристаллы, растворимые в воде, малорастворимые в спирте, нерастворимые в эфире. Температура плавления гистидина 287-288 0 С (с разл.).
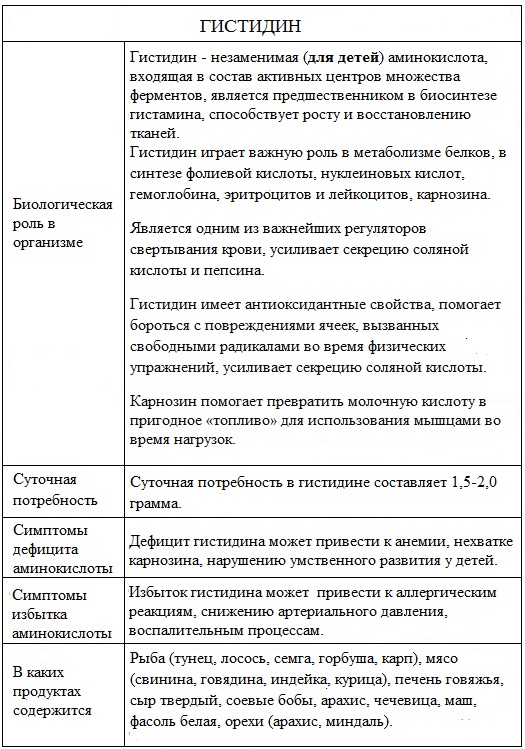
Биологическая роль
Гистидин входит в состав многих белков. Он принимает активное участие в синтезе карнозина (азотистого экстрактивного вещества мышц), улучшает азотистый баланс, функцию печени, повышает желудочную секрецию и моторику кишечника, иммунитет, нормализует сердечный ритм.
В значительном количестве содержится в гемоглобине, поэтому недостаток гистидина приводит к снижению уровня гемоглобина.
Недостаток или отсутствие гистидина замедляет синтез гемоглобина и приводит к развитию анемий в связи с тем, что белковая часть гемоглобина требует достаточно большого количества гистидина.
Гемоглобин является одним из резервов гистидина в организме и при недостатке гистидина происходит повышенное разрушение гемоглобина, в результате которого высвобождается гистидин.
При декарбоксилировании гистидина образуется гистамин. В ряде продуктов при их хранении, например в рыбе и сыре, происходит микробиологическое декарбоксилирование гистидина с образованием и накоплением больших количеств гистамина, что может иметь клинические последствия.
Между обменом гистидина и гистамина существует тесная связь.
Гистамин – биологически активный амин, который был синтезирован в 1907 г., позднее был изолирован из тканей млекопитающих. Гистамин (2- (4-имидазолил)этиламин) присутствует в растительных и животных тканях, является компонентом некоторых ядов и секретов, обладающих раздражающим действием.
Гистамин в организме обычно находится в неактивном состоянии. При некоторых патологических состояниях: аллергии, ожоги, обморожения, попадание в организм химических веществ (в том числе и лекарственных препаратов) гистамин накапливается в организме в значительных количествах и выделяется в свободном виде, который оказывает действие на окружающие ткани.
Свободный гистамин высокоактивен: вызывает спазм гладкой мускулатуры, расширяет кровеносные сосуды и увеличивает секрецию желудочного сока (стимулирует секрецию соляной кислоты и пепсина в желудке), снижает артериальное давление и частоту сердечных сокращений, принимает участие в развитии воспалительного процесса.
Природные источники
Гистидином богаты такие продукты как рыба (тунец, лосось, семга, горбуша, карп), мясо (свинина, говядина, индейка, курица), печень говяжья, сыр твердый, соевые бобы, арахис, чечевица, маш, фасоль белая, орехи (арахис, миндаль).
Области применения
Хлороводородная соль гистидина в медицинской практике применяется при язвенной болезни желудка и двенадцатиперстной кишки, гастритах, гепатитах, атеросклерозе, сниженном иммунитете.
1. Применяются при аллергических состояниях различного происхождения, при бронхиальной астме (димедрол, тавегил, супрастин, дипразин, фенкарол, диазолин).
2. Применяются для уменьшения секреции соляной кислоты при язвенной болезни желудка и двенадцатиперстной кишки (циметидин, ранитидин, фамотидин, низатидин),
ЦВЕТНЫЕ И ИМЕННЫЕ КАЧЕСТВЕННЫЕ РЕАКЦИИ НА БЕЛКИ
История химии в школьном курсе
Белки – основа жизнедеятельности любого организма на Земле. Это сложные высокомолекулярные природные соединения. Мономерами белков являются аминокислоты. Умение определять аминокислоты важно и в теоретическом, и в практическом аспекте. Определение аминокислот сопровождается написанием уравнений качественных реакций, что способствует углублению знаний по органической химии. Это умение имеет большое значение при заболеваниях, связанных с ослаблением иммунной системы людей (аллергические заболевания, нарушение функционирования ферментативных систем и т. д.), при которых основную роль играют белки. В данной ситуации необходимо оперативно и грамотно определять аминокислоты (белки).
Мы постарались в этой работе выяснить химизм качественных реакций на аминокислоты, указать роль отдельных ученых, в том числе российских, в исследовании белков, а также:
1) изучить и систематизировать имеющиеся литературные данные по качественным реакциям на белковые аминокислоты, составить базу данных о качественных, в том числе цветных, реакциях на белки; 2) научиться практически осуществлять качественные реакции; 3) выделить качественные реакции на белковые аминокислоты, изучаемые в школьном курсе химии.
Для аминокислот, постоянно встречающихся в составе белков, разработано множество цветных (в том числе именных) реакций. Многие из них высокоспецифичны, что позволяет определять ничтожные количества той или иной аминокислоты.
Надо помнить, что все качественные реакции – это реакции не собственно на белки, а на определенные аминокислоты, входящие в их состав.
Основной структурной единицей белков служат a -аминокислоты. В состав большинства природных белков входит около 20 a -аминокислот.
Общая формула белковых a -аминокислот:
Основным источником a -аминокислот для живого организма являются пищевые белки. Некоторые белковые аминокислоты синтезируются и самим организмом. Их называют заменимыми аминокислотами. Другие a -аминокислоты, необходимые для синтеза белков, синтезироваться в организме не могут и должны поступать только извне. Такие аминокислоты называют незаменимыми.
Все a -аминокислоты, входящие в состав белков, за исключением глицина (аминоуксусная кислота), содержат один или два асимметрических атома углерода и являются оптически активными соединениями. Они существуют в виде пар зеркальных изомеров (энантиомеры, или оптические антиподы), различающихся положением аминогруппы у асимметрического (хирального) атома углерода (он обозначен звездочкой). Расположение аминогруппы справа в проекционной формуле Фишера соответствует D-конфигурации, ее расположение слева – L-конфигурации:
Большинство D-изомеров обладает сладким вкусом, а L-изомеры – горькие или безвкусные.
В состав природных белков входят только L-аминокислоты. a -Аминокислоты D-ряда называют иногда неприродными, т. к. они не используются для построения белков человеческого организма.
a -Аминокислоты представляют собой кристаллические вещества, растворимые в воде, имеющие сравнительно высокую температуру плавления (200–300 °С). Способность a -аминокислот растворяться в воде является важным фактором в обеспечении их биологического функционирования. С нею связана всасываемость a -аминокислот, их транспорт в организме.
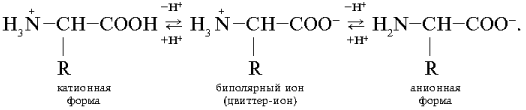
Такие аминокислоты имеют две ионизируемые группы: карбоксильную (–СООН) и аминогруппу (–NН2). В твердом состоянии и в водных растворах при определенных значениях рН среды a -аминокислоты существуют в виде биполярных ионов (цвиттер-ионы), представляющих собой внутреннюю соль. В биполярном ионе карбоксильная группа диссоциирована (–СОО – ) , а аминогруппа протонирована Ионизация молекул a -аминокислот зависит от рН раствора:
a -Аминокислоты содержат две различные функциональные группы: амино- и карбоксильную группы. Следовательно, это гетерофункциональные соединения.
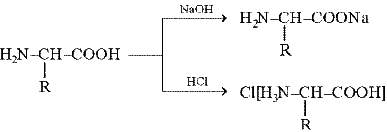
Аминогруппа обусловливает основные свойства вещества, а карбоксильная – кислотные, именно поэтому a -аминокислоты являются амфотерными соединениями, т. е. образуют соли как с кислотами, так и со щелочами:
Кроме того, a -аминокислоты могут вступать в другие химические реакции, характерные для амино- и карбоксильных групп.
Реакция Адамкевича (Адамкевича–Гопкинса (1874))
Описание опыта. К 2 мл 1%-го раствора триптофана приливают
1 мл концентрированной уксусной кислоты, встряхивают и по стенке пробирки осторожно добавляют
2 мл концентрированной серной кислоты. На границе двух жидкостей наблюдают образование красно-фиолетового окрашивания. При встряхивании жидкость окрашивается в фиолетовый цвет.
Реакция Ван Слайка
Это реакция определения первичной аминогруппы в алифатических аминах. a -Аминокислоты, содержащие первичную аминогруппу, реагируют с азотистой кислотой. При этом образуется неустойчивое диазосоединение, разлагающееся с выделением свободного азота и образованием a -гидроксикарбоновых кислот:
Реакция используется для количественного определения аминокислот по объему выделившегося газообразного азота.
Описание опыта. В пробирку наливают 1 мл 1%-го раствора глицина и равный объем 5%-го раствора нитрита натрия. Добавляют 0,5 мл концентрированной уксусной кислоты и осторожно взбалтывают смесь. Наблюдается выделение пузырьков газа:
Реакция Вуазена
Описание опыта. Если к 1 мл 5%-го раствора белка в 30%-м растворе едкого калия прибавить одну каплю 1,25%-го раствора формальдегида, 10 мл концентрированной соляной кислоты и через 10 минут прибавить 5–7 капель 0,05%-го раствора нитрита натрия, то появляется фиолетовое окрашивание, обусловленное присутствием в белке триптофана.
РЕАКЦИЯ МИЛЛОНА
Это реакция на аминокислоту тирозин. Реактив Миллона (раствор HgNO3 и Hg(NO2)2 в разбавленной HNO3, содержащей примесь HNO2) взаимодействует с тирозином с образованием ртутной соли нитропроизводного тирозина, окрашенной в розовато-красный цвет:
Описание опыта. К 2 мл концентрированного раствора тирозина прибавляют
1 мл реактива Миллона, встряхивают и осторожно нагревают пробирки на пламени спиртовки. Образуется красное окрашивание.
Реакция Гопкинса–Коле
Это реакция на аминокислоту триптофан.
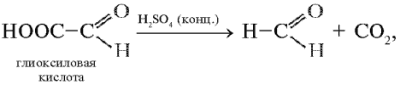
Описание опыта. 1 мл 0,005%-го раствора триптофана смешивают с равным объемом глиоксиловой кислоты НС(О)СООН* и к смеси прибавляют 10 капель 0,04 М раствора сульфата меди(II). Затем небольшими порциями (по несколько капель) добавляют 2–3 мл концентрированной серной кислоты, охлаждая пробирку после приливания очередной порции кислоты током холодной воды (или в ванночке со льдом). Полученную смесь оставляют на 10 мин при комнатной температуре, после чего ставят на 5 мин в кипящую водяную баню. Наблюдается образование сине-фиолетового окрашивания.
В этой реакции из глиоксиловой кислоты под действием концентрированной серной кислоты сначала получается формальдегид:
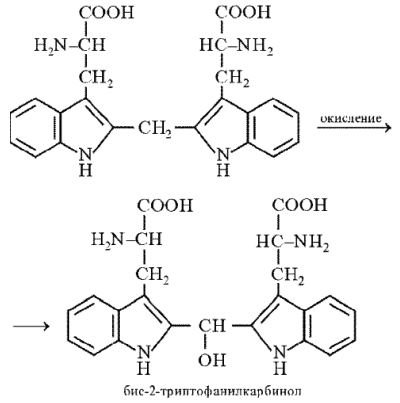
который затем конденсируется с триптофаном:
Продукт конденсации окисляется до бис-2-триптофанилкарбинола, который в присутствии минеральных кислот образует соли, окрашенные в сине-фиолетовый цвет:
Реакция Мак-карти и салливана
Это реакция на аминокислоту метионин.
Описание опыта. К 5 мл 0,02 н. раствора метионина прибавляют при перемешивании сначала 1 мл 14,3 н. раствора гидроксида натрия, а затем 0,3 мл свежеприготовленного 10%-го раствора нитропруссида натрия. Смесь 10 мин нагревают на водяной бане при 35–40 °С, затем в течение 2 мин охлаждают в ледяной воде. К смеси добавляют при помешивании 5 мл смеси соляной и фосфорной кислот. Полученный раствор взбалтывают 1 мин и охлаждают водой комнатной температуры в течение 10 мин. Образуется яркая красно-фиолетовая окраска.
Реакция Паули (Диазореакция Паули)
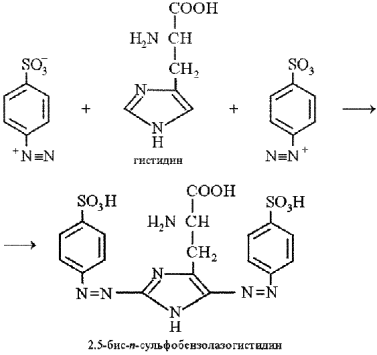
Эта реакция на аминокислоту гистидин основана на взаимодействии гистидина с диазобензолсульфоновой кислотой с образованием соединения вишнево-красного цвета.
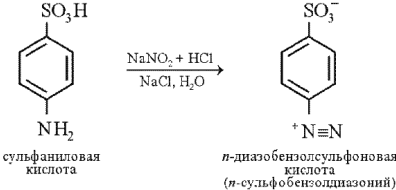
Реакцию диазотирования осуществляют при взаимодействии кислого раствора сульфаниловой кислоты с нитритом натрия. При этом образуется диазобензолсульфоновая кислота:
Эта кислота, взаимодействуя с гистидином, дает соединение вишнево-красного цвета:
Описание опыта. В пробирку наливают 1 мл 1%-го раствора сульфаниловой кислоты в 5%-м растворе соляной кислоты. Затем прибавляют 2 мл 0,5%-го раствора нитрита натрия, сильно встряхивают и немедленно приливают 2 мл 0,01%-го раствора гистидина. После перемешивания содержимого пробирки сразу приливают 6 мл 10%-го раствора соды. Появляется интенсивная вишнево-красная окраска.
Окончание следует
*К 2 г порошка магния (слегка увлажненного) прилить при охлаждении 50 мл (заранее охлажденного до 0 °С) насыщенного раствора щавелевой кислоты. Полученный осадок оксалата магния отфильтровывают и декантируют небольшим количеством воды. Фильтрат подкисляют уксусной кислотой и доводят до объема 200 мл (полученный раствор хранить в холодильнике!).



































 H3N + Н Н NH3 +
H3N + Н Н NH3 + 4. Напишите уравнение реакции гистидина с азотистой кислотой.
4. Напишите уравнение реакции гистидина с азотистой кислотой.
 Гистидин — 2-амино-3-имидазолилпропановая или α-амино-β-имидазолилпропионовая кислота.
Гистидин — 2-амино-3-имидазолилпропановая или α-амино-β-имидазолилпропионовая кислота.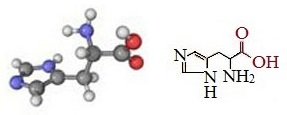

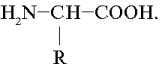
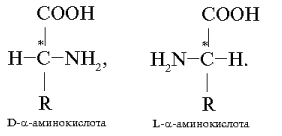
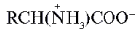 (цвиттер-ионы), представляющих собой внутреннюю соль. В биполярном ионе карбоксильная группа диссоциирована (–СОО – ) , а аминогруппа протонирована
(цвиттер-ионы), представляющих собой внутреннюю соль. В биполярном ионе карбоксильная группа диссоциирована (–СОО – ) , а аминогруппа протонирована 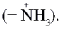 Ионизация молекул a -аминокислот зависит от рН раствора:
Ионизация молекул a -аминокислот зависит от рН раствора: