Реальные газы. Уравнение Ван-дер-Ваальса. Критическое состояние.
Реальным называется газ, между молекулами которого действуют силы межмолекулярного взаимодействия, состоящие из сил притяжения и сил отталкивания.
Для получения уравнения состояния реального газа необходимо учесть собственный объем молекул и энергию взаимодействия молекул на расстоянии. Наличие собственного объема молекул приводит к уменьшению объема, предоставленного молекулам, на некоторую величину. Силы притяжения между молекулами газа вызывают уменьшение давления молекул газа на стенки сосуда на некоторую величину рi.
Это уравнение может получено путем соответствующего изменения уравнения Менделеева-Клапейрона путем внесения в него поправок.
Уравнение состояния реального газа (уравнение Ван-дер-Ваальса) для одного моля имеет вид:
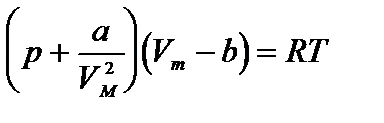 ,
,
где р — давление, оказываемое на стенки сосуда, VМ – объем одного моля газа, а и b — постоянные Ван-дер-Ваальса, имеющие для разных газов различные значения, определяемые опытным путем. Поправка 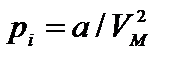 – внутреннее давление, обусловленное силами взаимного притяжения между молекулами. Поправка b характеризует ту часть объем, которая недоступна для движения молекул. Она равна учетверенному собственному объему молекул, содержащихся в моле газа:
– внутреннее давление, обусловленное силами взаимного притяжения между молекулами. Поправка b характеризует ту часть объем, которая недоступна для движения молекул. Она равна учетверенному собственному объему молекул, содержащихся в моле газа:
b= 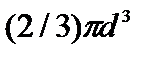 NA.
NA.
Уравнение Ван-дер-Ваальса для произвольной массы газа имеет вид:
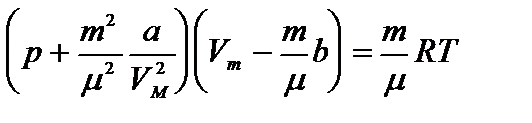
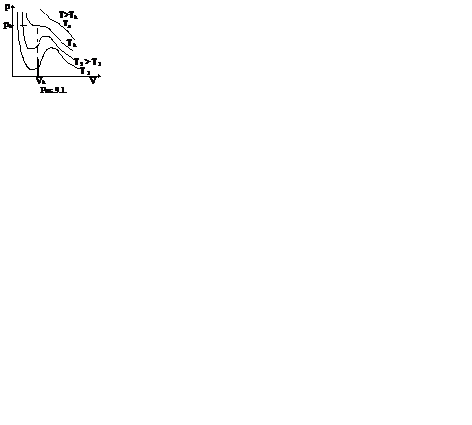
Уравнение Ван-дер-Ваальса позволяет построить теоретические изотермы реального газа и сравнить их с изотермами идеального газа и экспериментальными изотермами реального газа.
Уравнение Ван-дер-Ваальса после нескольких преобразований можно записать в виде:
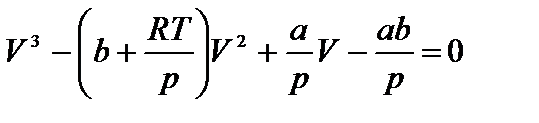 .
.
Это уравнение третьей степени относительно V. Кубическое уравнение может иметь либо три вещественных корня, либо один вещественный и два мнимых.
Первому случаю соответствуют изотермы при низких температурах – кривые для Т1 и Т2 (рис.9.1.) Второму случаю изотермы при высоких температурах (одно значение объема V отвечает одному значению давления р), то есть любая изотерма начиная от изотермы для Тк.
Совпадение изотерм идеального и реального газа наблюдается при малых давлениях и больших объемах (так как при этих условиях газ можно считать идеальным). Для семейства изотерм Ван-дер-Ваальса характерно так называемой критической изотермы (при температуре Тк) имеющий точку перегиба при некотором давлении рк и объеме Vк; при Т>Тк все изотермы идут монотонно, при Т
Уравнение Ван-дер-Ваальса описывает не только свойства газов и паров, но и жидкостей. Анализ изотерм реального газа показывает, что превращение реального газа в жидкость возможно только при температурах, меньших критической, и при соответствующих давлениях.
Дата добавления: 2015-04-01 ; просмотров: 18364 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Лекция № 7 Физические процессы в реальных газах

Физические процессы в реальных газах
Выписка из рабочей программы дисциплины «Молекулярная физика и термодинамика»
Название темы, литература
4.1 Физические процессы в реальных газах
Изотермы реального газа. Критическое состояние. Понятия пересыщенного пара, растянутой жидкости и метастабильного состояния. Внутренняя энергия реального газа. Эффект Джоуля — Томсона. Сжижение газов.
1. Изотермы Ван-дер-Ваальса
Уравнение состояния идеального газа (уравнение Менделеева-Клапейрона) имеет вид:
 .
.
Если в идеальном газе процесс протекает при постоянной температуре (изотермический процесс), то график зависимости  имеет вид (рис. 1):
имеет вид (рис. 1):

Рис. 1 Изотермы идеального газа
Уравнение состояния реального газа (уравнение Ван-дер-Ваальса) имеет вид:
 .
.
Для простоты предположим, что имеется 1 моль реального газа. Тогда уравнение Ван-дер-Ваальса можно записать в виде:
 .
.
 .
.
Приведём к общему знаменателю:
 ,
,
 .
.
Разделим на  :
:
 .
.
Сгруппируем члены с одинаковыми показателями степени объёма:
 . (1)
. (1)
Уравнение (1) является уравнением третьей степени и должно иметь три корня: V1, V2, V3. Это означает, что эти три значения объёма соответствуют одному значению давления и температуры.
Теоретические изотермы Ван-дер-Ваальса. Изотермы реальных газов.
 Из вида уравнений Ван-дер-Ваальса следует, что оно – третьей степени относительно величины V. Поэтому вид графиков достаточно сложный.
Из вида уравнений Ван-дер-Ваальса следует, что оно – третьей степени относительно величины V. Поэтому вид графиков достаточно сложный.
Изотерма, представляющая собой одно из решений уравнения Ван-дер-Ваальса.
Поэтому, в отличие от изотерм идеального газа, представляющих собой гиперболу, изотермы Ван-дер-Ваальса представляют собой кривую, являющуюся решением кубического уравнения, имеет максимум и минимум, а одному значению р1 соответствуют три возможных значения объёма V.
Теоретические изотермы для углекислого газа при различных температурах шкалы Цельсия (температуры указаны над  графиками). По оси абсцисс указан удельный объём – объем, приходящийся на единицу массы.
графиками). По оси абсцисс указан удельный объём – объем, приходящийся на единицу массы.
Из кривых видно, что по мере повышения температуры изотермы располагаются выше, что естественно, кроме того, максимумы и минимумы изотерм сближаются как по оси абсцисс (уменьшается разность между отвечающими им объемами), так и по оси ординат (уменьшается соответствующая разность давлений). Наконец, при вполне определенной критической температуре (на рис. при 31,4° С) максимум и минимум изотермы сливаются, вырождаясь в точку перегиба при значении n = 2,7 л/моль и р = 73 атм. Это значит, что при повышении температуры постепенно уменьшается различие между тремя значениями объема, соответствующими одному и тому же значению давления, т. е. уменьшается различие между тремя корнями уравнения Ван-дер-Ваальса.
При определенной (критической) температуре Тк все три значения объема сливаются, корни уравнения становятся кратными. При этой температуре исчезает, следовательно, различие между различными состояниями вещества. Это, очевидно, и есть критическая температура, существование которой является характерным свойством вещества.
При температуре выше критической поведение реального газа приближается к поведению идеального, описываемого законом Бойля-Мариотта.
Рассмотрим теперь экспериментальные данные для зависимости p=f(V), получаемые по простой схеме.

Рис. а) схема опыта (М-манометр для измерения давления), б – график полученного результата.
Оказалось, что газовое состояние существовало лишь на участке 1-2, на участке 2-3 вещество представляло собой двухфазную систему «пар+жидкость», а на участке 3-4 наблюдается сжатие практически несжимаемой жидкости.
Более тщательное исследование газов и жидкостей, очищенных от примесей, пыли и т. п. показали, что из теоретической изотермы можно реализовать некоторые участки.
 Сравнение теоретической и реальной изотерм Ван-дер-Ваальса.
Сравнение теоретической и реальной изотерм Ван-дер-Ваальса.
На участке cd изотермы вещество находится в неустойчивом «метастабильном» состояния пересыщенного пара, которое при малом внешнем воздействии самопроизвольно конденсируется в жидкость в виде капель тумана. Это состояние используется, в частности, в камерах Вильсона для наблюдения движения микрочастиц.
На участке af изотермы вещество представляет собой «перегретую жидкость» – также метастабильное состояние, самопроизвольно стремящееся в парообразное состояние. Это состояние также используется для наблюдения за микрочастицами, в пузырьковых камерах.
Участок fd не реализуется ни в каких известных опытах.
На рис. представлены графики для углекислого газа , полученные реально, а не расчетным путем.
, полученные реально, а не расчетным путем.
Фазовая диаграмма для углекислого газа.
Видно, что при температурах ниже Тк на каждой изотерме имеется горизонтальный участок ВС, вдоль которого постоянна не только температура, но и давление, а молярный объём может принимать любые значения от VB до VC.
Оказывается, на участке АВ вещество – жидкость, а на участке СТ – углекислота находится в газообразном состоянии. На участке СВ углекислота одновременно находится в двух агрегатных состояниях — жидком и газообразном. Точка С соответствует началу конденсации СО а при изотермическом сжатии, а точка В — концу конденсации. Наоборот, при изотермическом расширении жидкой углекислоты точка В соответствует началу кипения, а точка С — его концу. Следовательно, точка В соответствует состоянию кипящей жидкости, а точка С состоянию так называемого сухого насыщенного пара. В произвольном состоянии М области ВС СО2 представляет собой смесь кипящей жидкости и сухого насыщенного пара. Такая смесь называется влажным паром.
Обнаружено, что при T>Tк переход в жидкое состояние невозможен ни при каких степенях сжатия.
Параметры критической точки вычислены и равны:

где a и b – константы уравнения Ван-дер-Ваальса, R – универсальная газовая постоянная.
Для СО2: Ткр = 304К; ркр = 7,38 МПа; 
Фазовая диаграмма четко делится на ряд областей.
I — первая область — область газа — ни при каких степенях сжатия газа переход в жидкое и твердое состояние невозможен.
II – «газ – пар» — происходит сжатие, которое начиная с некоторого значения р, приводит к возникновению двухфазного состояния
III — двухфазное состояние: «жидкость + пар», количество газообразной и жидкой фаз может быть найдено «правилом рычага», МС – доля жидкой фазы, МВ – доля паровой фазы.
IV — жидкость — практически несжимаемая материя.
13.3. Изотермы реального газа. Критическое состояние
Как мы уже отмечали, по мере увеличения объема или повышения температуры уравнение Ван-дер-Ваальса переходит в уравнение Клапейрона-Менделеева. Наоборот, c увеличением давления и понижением температуры различие между ними становится заметнее. Формально это связано с тем, что рассматриваемое нами уравнение состояния реального газа представляет собой алгебраическое уравнение третьей степени относительно  , в то время как для идеального газа это уравнение первой степени. Убедиться в этом можно, раскрыв скобки в уравнении Ван-дер-Ваальса для одного моля газа и сгруппировав подобные члены при одинаковых степенях . Получим новую форму записи этого уравнения[1] :
, в то время как для идеального газа это уравнение первой степени. Убедиться в этом можно, раскрыв скобки в уравнении Ван-дер-Ваальса для одного моля газа и сгруппировав подобные члены при одинаковых степенях . Получим новую форму записи этого уравнения[1] :
 . (13.3)
. (13.3)
Давление  и температура
и температура  входят в это уравнение как параметры. Из теории алгебраических уравнений известно, что уравнение третьей степени должно иметь три корня (либо три действительных, либо один действительный и два комплексных). Применительно к уравнению (13.3) это три значения объема
входят в это уравнение как параметры. Из теории алгебраических уравнений известно, что уравнение третьей степени должно иметь три корня (либо три действительных, либо один действительный и два комплексных). Применительно к уравнению (13.3) это три значения объема  ,
,  и
и  . В зависимости от величины параметров
. В зависимости от величины параметров  и эти корни могут быть либо действительными равными (
и эти корни могут быть либо действительными равными ( ), либо действительными разными (
), либо действительными разными ( ), либо один из корней действительный, а два других – мнимые.
), либо один из корней действительный, а два других – мнимые.
Если в уравнении (13.3) зафиксировать значение температуры ( ), то оно будет соответствовать теоретической изотерме реального газа, которая изображена на рис. 124. Теоретическая изотерма изображена сплошной линией ABEFCD и называется изотермой Ван-дер-Ваальса.
), то оно будет соответствовать теоретической изотерме реального газа, которая изображена на рис. 124. Теоретическая изотерма изображена сплошной линией ABEFCD и называется изотермой Ван-дер-Ваальса.
Для анализа отдельных участков изотермы Ван-дер-Ваальса рассмотрим опыт, схема которого изображена на том же рисунке. Толстостенная стеклянная трубка, закрытая с одного конца поршнем, заполнена некоторым реальным газом и расположена параллельно оси абсцисс. К левому концу трубки прикреплен ртутный манометр. Объем газа указывается непосредственно положением поршня, а давление – высотой ртутного столба, параллельного оси ординат. Если при постоянной температуре перемещать поршень и регистрировать давление, то можно выяснить, насколько правильно уравнение Ван-дер-Ваальса отражает поведение реального газа[2].
На рис. 125 пунктиром изображен результат подобного опыта, соответствующий некоторой температуре . В соответствии с теорией при уменьшении объема давление газа возрастает (участок AB). При достижении объема  наступает отклонение от теории: давление перестает изменяться при уменьшении объема и остается постоянным. Реальный газ начинает конденсироваться, т. е. часть количества газа превращается в жидкость. Газ, который путем сжатия можно превратить в жидкость, называется паром. В трубке образуется жидкость, которая находится в равновесии со своим паром.
наступает отклонение от теории: давление перестает изменяться при уменьшении объема и остается постоянным. Реальный газ начинает конденсироваться, т. е. часть количества газа превращается в жидкость. Газ, который путем сжатия можно превратить в жидкость, называется паром. В трубке образуется жидкость, которая находится в равновесии со своим паром.  Равновесие надо понимать следующим образом: сколько молекул газа в единицу времени конденсируется, столько же молекул жидкости испаряется, образуя пар. Газ, находящийся в равновесии со своей жидкостью, называется насыщающим паром, а его давление – давлением насыщающего пара. Давление насыщающего пара зависит только от температуры. Поэтому, пока весь пар не превратится в жидкость, его давление не изменяется и равно давлению насыщающего пара. Таким образом, конденсация пара изображается на графике горизонтальной линией ВС. В точке С конденсация заканчивается. Весь насыщающий пар превращается в жидкость объемом
Равновесие надо понимать следующим образом: сколько молекул газа в единицу времени конденсируется, столько же молекул жидкости испаряется, образуя пар. Газ, находящийся в равновесии со своей жидкостью, называется насыщающим паром, а его давление – давлением насыщающего пара. Давление насыщающего пара зависит только от температуры. Поэтому, пока весь пар не превратится в жидкость, его давление не изменяется и равно давлению насыщающего пара. Таким образом, конденсация пара изображается на графике горизонтальной линией ВС. В точке С конденсация заканчивается. Весь насыщающий пар превращается в жидкость объемом  . Как известно, жидкость практически несжимаема, поэтом очень незначительное уменьшение объема может быть получено только при значительном увеличении давления. Поэтому линия CD, соответствующая жидкому состоянию, идет круто вверх. Полученная в результате опыта кривая называется опытной изотермой реального газа. Она содержит горизонтальный участок (называемый плато), отражающий конденсацию пара.
. Как известно, жидкость практически несжимаема, поэтом очень незначительное уменьшение объема может быть получено только при значительном увеличении давления. Поэтому линия CD, соответствующая жидкому состоянию, идет круто вверх. Полученная в результате опыта кривая называется опытной изотермой реального газа. Она содержит горизонтальный участок (называемый плато), отражающий конденсацию пара.
Теоретическая изотерма хорошо согласуется с опытной при всех давлениях, исключая область конденсации насыщающего пара. В этой области наблюдается сильное расхождение теории Ван-дер-Ваальса и эксперимента. Однако область «горбов» теоретической изотермы точно попадает на область конденсации пара и это позволяет качественно установить ряд важных закономерностей, наблюдаемых на опыте. Рассмотрим эту область изотермы подробнее.
Повторим рассмотренный выше опыт, но с модельным газом Ван-дер-Ваальса, который при данной температуре может быть сжат до объема меньшего, чем объем насыщающего пара (участок EB). В теоретической модели нет конденсации. Но сжатие реального газа в соответствии с используемой моделью оказывается возможным, если сжимаемый газ тщательно очистить от пыли. В этом случае можно наблюдать задержку возникновения конденсации. Состояние газа, соответствующее этому участку, называется пересыщенным паром. Это состояние неустойчиво. В пересыщенном паре спонтанно (т. е. самопроизвольно) может возникать конденсация, при которой давление скачком падает до значения насыщающего пара. Центрами конденсации являются обычно пылинки, поверхности тел и т. д. Поэтому спонтанная конденсация проявляется в образовании мельчайших капелек жидкости (тумана) или же запотевании поверхностей, например, стекол.
Аналогичным образом при изотермическом расширении жидкости можно наблюдать задержку парообразования. При этом давление в системе будет ниже давления насыщающего пара. Участок CF (см. рис. 125) теоретической кривой хорошо согласуется с задержкой парообразования. Эта часть изотермы Ван-дер-Ваальса соответствует неустойчивому состоянию растянутой жидкости. Иногда ее называют перегретой, так как она имеет объем, больший, чем это соответствует данной температуре.

При определенных условиях газ может находиться в неустойчивых состояниях (пересыщенного пара и перегретой жидкости) длительное время. Поэтому эти состояния называются метастабильными1. Метастабильными называют равновесные состояния с ограниченной устойчивостью. При отклонении от этого состояния система не стремится к нему обратно, а легко переходит в другое устойчивое состояние. Метастабильные состояния существуют ограниченное время.
Участок FE изотермы Ван-дер-Ваальса на практике не реализуется, так как он соответствует нереальному случаю роста давления при увеличении объема. Причина его существования лежит в ограниченности модели. Если этот участок мешал бы практическому использованию теории, модель была бы уточнена, но в данном случае этого не требуется. И хотя модель Ван-дер-Ваальса весьма груба, она все же позволяет сделать очень много. Во-первых, она уточняет опытную изотерму. С учетом возможных метастабильных состояний опытная изотерма в общем случае должна содержать «зубцы», имеющие разный знак для прямого и обратного хода процесса (сжатие и разряжение). Это показано на рис. 126.
Кроме этого, теория Ван-дер-Ваальса позволяет объяснить существование очень важного свойства реальных газов, называемое критическим состоянием. Рассмотрим этот вопрос подробнее.
По мере повышения температуры изотермы Ван-дер-Ваальса поднимаются вверх, а область «горбов» на них становится уже. Это означает, что уменьшается разность объемов  на опытных изотермах. Если рассмотреть семейство изотерм реального газа, то можно обратить внимание на то, что при некоторой температуре эта разность объемов становится равной нулю. Соответствующая ей изотерма не содержит ни «горбов», ни плато. На ней вместо горизонтального участка имеется перегиб. Такая изотерма отделяет опытные изотермы с горизонтальными участками от изотерм без горизонтальных участков. Она называется критической, а соответствующая ей температура критической температурой
на опытных изотермах. Если рассмотреть семейство изотерм реального газа, то можно обратить внимание на то, что при некоторой температуре эта разность объемов становится равной нулю. Соответствующая ей изотерма не содержит ни «горбов», ни плато. На ней вместо горизонтального участка имеется перегиб. Такая изотерма отделяет опытные изотермы с горизонтальными участками от изотерм без горизонтальных участков. Она называется критической, а соответствующая ей температура критической температурой  . Точке перегиба на критической изотерме соответствует критическое давление
. Точке перегиба на критической изотерме соответствует критическое давление  и критический объем
и критический объем  (рис. 127).
(рис. 127).
Величины , и называются критическими параметрами и являются индивидуальными характеристиками газа. Для температур выше вещество может существовать только в газообразном состоянии и никаким давлением его нельзя перевести в жидкое состояние. Чем выше температура по сравнению с критической, тем меньше различие между изотермами реального и идеального газов. Из сказанного становится очевидным, что давление насыщающего пара не может быть больше критического, так как при этих давлениях нет конденсации. По той же причине, объем образовавшейся жидкости меньше критического объема. (Здесь следует еще раз напомнить, что рассмотрение касается газа в количестве одного моля.)
В критической точке исчезает всякое различие между жидкостью и паром, а переход вещества из газообразного состояния в жидкое происходит непрерывно. Вблизи критической точки возрастают флуктуации плотности вещества, приводящие к сильному рассеянию света. Это явление получило название опалисценции (по имени минерала опала, также сильно рассеивающего свет). Значения критических параметров можно найти из уравнения Ван-дер-Ваальса. Для температур меньших критической уравнение (13.3) имеет три различных действительных корня. Для критической температуры уравнение должно иметь три одинаковых корня ( ). Поэтому, согласно теории алгебраических уравнений, уравнение Ван-дер-Ваальса для критической точки, записанное в форме (13.3), должно быть тождественно уравнению
). Поэтому, согласно теории алгебраических уравнений, уравнение Ван-дер-Ваальса для критической точки, записанное в форме (13.3), должно быть тождественно уравнению
 , (13.4)
, (13.4)
где – текущее значение объема.


Сравнивая коэффициенты при одинаковых степенях объема в уравнениях (13.3) и (13.4), найдем «координаты» критической точки, т. е. теоретические выражения для критических параметров:
 .
.
Критические параметры можно определить экспериментально, а из них рассчитать значения постоянных Ван-дер-Ваальса. Этим самым теоретическое уравнение состояния оказывается привязанным как к индивидуальным свойствам газов, так и к любой реальной ситуации.
Если соединить между собой точки, соответствующие  и
и  всех изотерм (рис. 128), то получим куполообразную кривую, ограничивающую область двухфазового состояния вещества (жидкость–газ). Здесь необходимо указать, что отличие пара от газа только в том, что газ никакими давлениями нельзя перевести в жидкость, а пар – можно.
всех изотерм (рис. 128), то получим куполообразную кривую, ограничивающую область двухфазового состояния вещества (жидкость–газ). Здесь необходимо указать, что отличие пара от газа только в том, что газ никакими давлениями нельзя перевести в жидкость, а пар – можно.
13.4. Внутренняя энергия реального газа. Эффект Джоуля-Томсона
Одним из важнейших отличий модели реального газа является учет сил межмолекулярного взаимодействия. Это накладывает отпечаток на величину внутренней энергии. Внутренняя энергия идеального газа представляет собой сумму кинетических энергий движения молекул и не зависит для данного количества газа ни от объема, ни от давления. Зависит она только от температуры. В реальном газе силы притяжения между молекулами играют существенную роль. Поэтому пренебрегать потенциальной энергией взаимодействия уже нельзя. Следовательно, внутренняя энергия реального газа должна складываться из кинетической энергии движения молекул и потенциальной энергии их взаимодействия:
 (13.5)
(13.5)
Потенциальная энергия должна зависеть от объема, так как при изменении объема изменяется расстояние между молекулами газа. Существование потенциальной энергии взаимодействия между молекулами накладывает отпечаток на изопроцессы, происходящие в реальных газах. Изотермический процесс был уже изложен выше. Теперь рассмотрим процесс адиабатического расширения реальных газов.
В соответствии с определением, при адиабатических процессах отсутствует обмен энергией с окружающей средой. Совершаемая при этом работа связана только с изменением внутренней энергии. Адиабатическое расширение идеального газа без совершения работы происходит при неизменном запасе внутренней энергии, т. е. без изменения температуры. В аналогичном случае для реального газа расширение приведет к изменению потенциальной энергии взаимодействия молекул, а условие неизменности внутренней энергии требует соответствующего изменения кинетической энергии. Изменение кинетической энергии означает изменение температуры реального газа. Для одного моля имеет место соотношение:
 (13.6)
(13.6)

Следовательно, при изменении объема реального газа без совершения работы против внешних сил и без теплообмена с окружающей средой должно наблюдаться изменение температуры. Первую попытку наблюдать явление такого рода предпринял Джоуль. Однако, чувствительность его установки была недостаточной. Несколько позже, на более чувствительной установке Джоулем совместно с Томсоном был экспериментально обнаружен эффект изменения температуры адиабатически расширяющегося (без совершения работы) газа, который получил название эффекта Джоуля–Томсона.
Сущность опыта Джоуля и Томсона состояла в следующем. В трубку, соединяющую два баллона с газом, помещалась пористая перегородка (дроссель). На рис. 129 изображен отрезок соединительной трубки с дросселем (баллоны с газом не показаны). По обе стороны от дросселя помещались чувствительные термометры. Давления  и
и  поддерживались постоянными. Условия опыта были таковы, что перепад давления при протекании газа по трубкам был только в месте нахождения пористой перегородки. Следовательно, в том же месте должно наблюдаться изменение температуры, которое и могло быть зафиксировано термометрами.
поддерживались постоянными. Условия опыта были таковы, что перепад давления при протекании газа по трубкам был только в месте нахождения пористой перегородки. Следовательно, в том же месте должно наблюдаться изменение температуры, которое и могло быть зафиксировано термометрами.
Опыт показал, что для большинства газов расширение сопровождается охлаждением, т. е. температура газа перед дросселем больше, чем после него. Для водорода знак эффекта противоположен.
Эффект Джоуля–Томсона принято называть положительным, если при адиабатическом расширении газ охлаждается. Если же газ нагревается, то эффект отрицательный.
Существование эффекта Джоуля–Томсона является следствием отступления свойств реальных газов от модели идеального газа, а знак его зависит от того, какая из поправок Ван-дер-Ваальса (  или
или  ) играет большую роль.
) играет большую роль.
Для установления связи знака эффекта Джоуля–Томсона с уравнением Ван-дер-Ваальса рассмотрим два предельных случая:
1) газ, для которого можно пренебречь поправкой ;
2) газ, для которого можно пренебречь поправкой .
Поправка в уравнении Ван-дер-Ваальса связана с наличием сил межмолекулярного взаимодействия. Поэтому в первом случае силами притяжения можно пренебречь и учитывать только силы отталкивания. Потенциальная энергия  сил отталкивания как функция расстояния между молекулами изображена на рис. 130а. Чем меньше давление газа, тем больше средние расстояния
сил отталкивания как функция расстояния между молекулами изображена на рис. 130а. Чем меньше давление газа, тем больше средние расстояния  между молекулами. Отсюда, как это следует из рис. 130 а, с уменьшением давления потенциальная часть внутренней энергии убывает:
между молекулами. Отсюда, как это следует из рис. 130 а, с уменьшением давления потенциальная часть внутренней энергии убывает:
 .
.
Следовательно, согласно выражению (13.6) температура газа увеличивается, т. е.  >0. Таким образом газ, для которого можно пренебречь поправкой в уравнении Ван-дер-Ваальса, при адиабатическом расширении нагревается.
>0. Таким образом газ, для которого можно пренебречь поправкой в уравнении Ван-дер-Ваальса, при адиабатическом расширении нагревается.
Второй случай  относится к газу, состоящему из очень маленьких по размеру молекул, так как – это поправка на объем молекул. Если размеры молекул очень малы, то силы отталкивания между ними не имеют существенной роли (за исключением моментов столкновений). Зависимость потенциальной энергии сил молекулярного притяжения от расстояния между
относится к газу, состоящему из очень маленьких по размеру молекул, так как – это поправка на объем молекул. Если размеры молекул очень малы, то силы отталкивания между ними не имеют существенной роли (за исключением моментов столкновений). Зависимость потенциальной энергии сил молекулярного притяжения от расстояния между

молекулами изображена на рис.130 б. Теперь при расширении газа потенциальная компонента внутренней энергии изменяется следующим образом:
 .
.
(Знак минус перед скобками в этом выражении соответствует тому, что потенциальная энергия отрицательна)
Следовательно, в данном случае знак эффекта меняется, т. е.
Уравнение ван дер ваальса и его изотермы
Рис. 3.7.2. Зависимость сил притяжения и сил отталкивания от расстояния
Рис. 3.7.3. Зависимость потенциальной энергии от расстояния
Глубина потенциала равна . при . (расстояние, соответствующее наибольшей энергии связи молекул). Отметим, что в данном потенциале не учтены ориентационные взаимодействия, существенные для многоатомных молекул и кристаллов.
Учитывая совместное действие сил притяжения и сил отталкивания и полученные поправки для объема и давления в уравнении Менделеева — Клапейрона, получим уравнение Ван-дер-Ваальса для реального газа
(P + ν 2 a/V 2 )(V-νb) = νRT (3.7.3)
или для одного моля —
Данное уравнение справедливо при условии νb 2 a/V 2 2 и раскрыв скобки, получим
http://pandia.ru/text/80/172/23046.php
http://www.chem-astu.ru/chair/study/physics-part1/?p=201