ИЗОТЕРМА ВАНТ — ГОФФА, ВЫВОД И АНАЛИЗ
Константа равновесия определяют условия равновесия, когда концентрация (парциальные давления) является равновесными. В каком направлении пойдёт химическая реакция, если парциальное давление исходных веществ и продуктов реакции отличаются от равновесных? Ответить на этот вопрос поможет уравнение изотермы химической реакции.
Рассмотрим уравнение реакции: aA + bB ↔ cC + dD
Для изобарно-изотермического процесса изменение Гиббса равно:
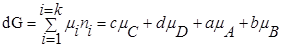
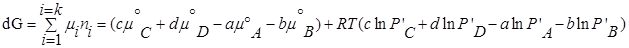 P’- неравновесное, парциальное давление компонентов.
P’- неравновесное, парциальное давление компонентов.
где 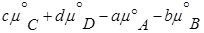 =
= 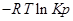
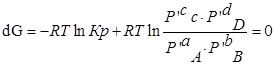
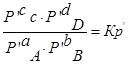
 — по форме записи напоминает константу равновесия, но отличается тем, что вместо равновесных давлений в него входят величины давления в данный момент времени.
— по форме записи напоминает константу равновесия, но отличается тем, что вместо равновесных давлений в него входят величины давления в данный момент времени.
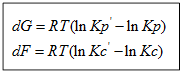
( 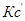 ) — это выражение, куда входят величины концентраций в данный момент времени.
) — это выражение, куда входят величины концентраций в данный момент времени.
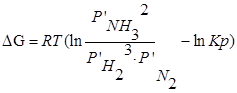
АНАЛИЗ ИЗОТЕРМЫ ВАНТ-ГОФФА
1. Главное значение изотермы реакции состоит в том, что она позволяет рассчитать ∆Gили ∆Fобратимый реакции для заданного состава реакционной смеси и определить, в каком направлении, и до какого предела протекает реакция при известных условиях.
а) Если Кр˃Кр’, то lnКр˃lnКр’; ∆G˂0- реакция идет самопроизвольно в прямом направлении.
б) Если Кр˂Кр’, то lnКр˂lnКр’; ∆G˃0- реакция протекает самопроизвольно в обратном направлении, в сторону образования продуктов.
в) Если Кр=Кр’, то lnКр=lnКр’; ∆G=0- равновесие.
Если парциальное давление всех участников реакции в данный момент времени равны атмосферному давлению
Пусть PA’=PB’=PC’=PD’=1( 1,013*10 5 Па), тоKp’=1; Тогда логарифм этого выражения будет равен нулю (lnKp’=0), а уравнение изотермы Вант – Гоффа примет вид:
∆G⁰=RTlnKp-стандартная энергия Гиббса
Выразм константу равновесия из последнего уравнения и получим:
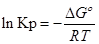
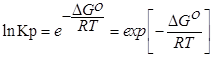
 Пример решения задачи:
Пример решения задачи:
В объеме 10л, взяли: 320(г) О2 , 10(г) Н2 и 180(г) паров воды . Определите, в какую направлении пойдёт химическая реакция: 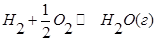 , если при температуре Т
, если при температуре Т
(Кс=10). Процесс изохорный.
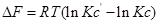
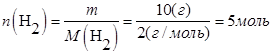 ;
;
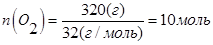 ;
;
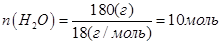 ;
;
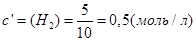 ;
;
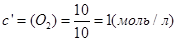 ;
;
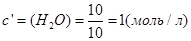 ;
;
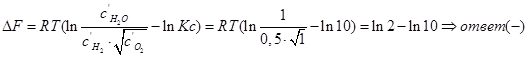
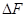 ˂0 — реакция идет в прямом направлении.
˂0 — реакция идет в прямом направлении.
ВЛИЯНИЕ ВНЕШНИХ УСЛОВИЙ НА КОНСТАНТУ
РАВНОВЕСИЯ. УРАВНЕНИЕ ИЗОБАРЫ И ИЗОХОРЫ ВАНТ-ГОФФА (В-Г)
Константы равновесия — это величины постоянные при данной температуре. При изменении температуры константа равновесия изменяется, и довольно существенно.
Изменение константы равновесия и направления химической реакции в зависимости от температуры количественно характеризует уравнение изобары изохоры химической реакции.
ВЫВОД УРАВНЕНИЯ ИЗОБАРЫ И ИЗОХОРЫ
Разделим уравнение изотермы Вант- Гоффа на температуру:
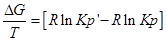
Продифференцируем его по Т и перепишем:
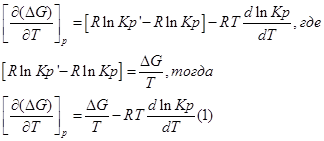
Представим уравнение Гиббса – Гельмгольца в виде:
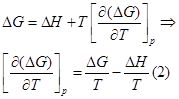
Из уравнения (1) вычтем уравнение (2):
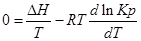
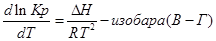
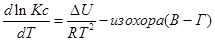
Эти уравнения показывают влияния температуры на константу равновесия, где определяющим фактором является тепловой эффект химической реакции.
Влияние температуры на константу равновесия определяется типом реакци.
1. Если тепловой эффект реакци ∆H(∆U)˃0(эндотермическая, поглощение), то 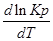 ˃0, тогда при увеличении температуры (Т↑) константа равновесия Кр увеличивается.
˃0, тогда при увеличении температуры (Т↑) константа равновесия Кр увеличивается.
В обратном — Т↓,Кр↓.
2. Если ∆H(∆U)˂0 (экзотермическая, выделение), то ˂0, тогда при повышении температуры константа равновесия Кр уменьшается или Кр увеличивается при понижении температуры.
В обратном — Т↑,Кр↓.
3. Если ∆H(∆U)=0 , тов этом случае константа равновесия не зависит от температуры Кp ≠ f(T).
ИНТЕГРИРОВАНИЕ ИЗОБАРЫ В-Г
1. Приближенное интегрирование ∆Н ≠ f(Т),
тогда 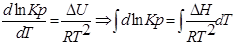 ;
;
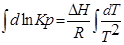 ;
;
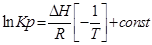 ;
;
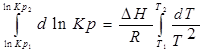 ;
;
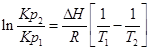 ;
;
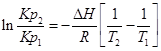 .
.
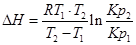
С помощью этого уравнения можно найти:
1. ∆Н (тепловой эффект реакции), если известны равновесия при двух различных температурах (Кр1(Т1) и Кр2(Т2))
2. Кр2(Т2) – константу равновесия при температуре Т2, если известна константа равновесия при другой температуре и тепловой эффект реакции (Кр1(Т1) и ∆Н).
Так как после интегрирования мы получили уравнение прямой, то эта зависимость может быть представлена на графике: lnKp(1) lnKp(2)
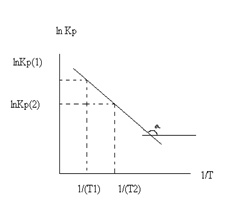
Тангенс угла наклона прямой реакции, исходя из уравнения прямой:
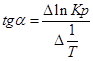 ;.
;.
Зависимость теплового эффекта от температуры выражается уравнением:
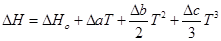
Подставим это уравнение в уравнение изобары Вант- Гоффа:
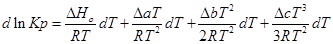 ;
;
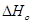 при Т=0(К)
при Т=0(К)
Проинтегрируем это уравнение и получим:
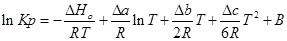 ;
;
где В — постоянная интегрирования, для нахождения необходимо знать значения константы равновесия Кр при любой фиксированной температуре.
Однако проводить расчеты с использованием данного уравнения довольно сложно и используется довольно редко.
Пример. Определим изменение эффекта реакции Fe+H2O+FeO+H2, если для Т1=900К, Кр1=1,452, а для Т2=1025К Кр2=1,285.
Используем уравнение: 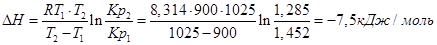
При повышении температуры от 900 до 1025К выделится дополнительно 7,5 кДж/моль теплоты.
| | | следующая лекция ==> | |
| Метод статистического моделирования нагрузки на ЭВМ | | | Строение и характеристика макроэргических соединений на примереАТФ |
Дата добавления: 2016-02-02 ; просмотров: 9784 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Уравнение вант гоффа энергия гиббса
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Конспект лекций для студентов биофака ЮФУ (РГУ)
1.7 ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Как было показано выше, протекание самопроизвольного процесса в термодинамической системе сопровождается уменьшением свободной энергии системы (dG 2 Y > 0. Таким образом, условием термодинамического равновесия в закрытой системе является минимальное значение соответствующего термодинамического потенциала :
Изобарно-изотермические (P = const, T = const):
Изохорно-изотермические (V = const, T = const):
Состояние системы с минимальной свободной энергией есть состояние термодинамического равновесия:
Термодинамическим равновесием называется такое термодинамическое состояние системы, которое при постоянстве внешних условий не изменяется во времени, причем эта неизменяемость не обусловлена каким-либо внешним процессом.
Учение о равновесных состояниях – один из разделов термодинамики. Далее мы будем рассматривать частный случай термодинамического равновесного состояния – химическое равновесие. Как известно, многие химические реакции являются обратимыми, т.е. могут одновременно протекать в обоих направлениях – прямом и обратном. Если проводить обратимую реакцию в закрытой системе, то через некоторое время система придет в состояние химического равновесия – концентрации всех реагирующих веществ перестанут изменяться во времени. Необходимо отметить, что достижение системой состояния равновесия не означает прекращения процесса; химическое равновесие является динамическим, т.е. соответствует одновременному протеканию процесса в противоположных направлениях с одинаковой скоростью. Химическое равновесие является подвижным – всякое бесконечно малое внешнее воздействие на равновесную систему вызывает бесконечно малое изменение состояния системы; по прекращении внешнего воздействия система возвращается в исходное состояние. Ещё одним важным свойством химического равновесия является то, что система может самопроизвольно прийти в состояние равновесия с двух противоположных сторон. Иначе говоря, любое состояние, смежное с равновесным, является менее устойчивым, и переход в него из состояния равновесия всегда связан с необходимостью затраты работы извне.
Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации С, парциальные давления P или мольные доли X реагирующих веществ. Для некоторой реакции

соответствующие константы равновесия выражаются следующим образом:
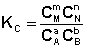 (I.78)
(I.78) 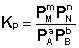 (I.79)
(I.79)
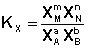 (I.80)
(I.80)
Константа равновесия есть характерная величина для каждой обратимой химической реакции; величина константы равновесия зависит только от природы реагирующих веществ и температуры. Выражение для константы равновесия для элементарной обратимой реакции может быть выведено из кинетических представлений.
Рассмотрим процесс установления равновесия в системе, в которой в начальный момент времени присутствуют только исходные вещества А и В. Скорость прямой реакции V1 в этот момент максимальна, а скорость обратной V2 равна нулю:
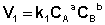 (I.81)
(I.81)
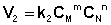 (I.82)
(I.82)
По мере уменьшения концентрации исходных веществ растет концентрация продуктов реакции; соответственно, скорость прямой реакции уменьшается, скорость обратной реакции увеличивается. Очевидно, что через некоторое время скорости прямой и обратной реакции сравняются, после чего концентрации реагирующих веществ перестанут изменяться, т.е. установится химическое равновесие.
Приняв, что V1 = V2, можно записать:
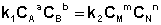 (I.83)
(I.83)
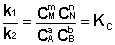 (I.84)
(I.84)
Т.о., константа равновесия есть отношение констант скорости прямой и обратной реакции. Отсюда вытекает физический смысл константы равновесия: она показывает, во сколько раз скорость прямой реакции больше скорости обратной при данной температуре и концентрациях всех реагирующих веществ, равных 1 моль/л.
Теперь рассмотрим (с некоторыми упрощениями) более строгий термодинамический вывод выражения для константы равновесия. Для этого необходимо ввести понятие химический потенциал . Очевидно, что величина свободной энергии системы будет зависеть как от внешних условий (T, P или V), так и от природы и количества веществ, составляющих систему. В случае, если состав системы изменяется во времени (т.е. в системе протекает химическая реакция), необходимо учесть влияние изменения состава на величину свободной энергии системы. Введем в некоторую систему бесконечно малое количество dni молей i-го компонента; это вызовет бесконечно малое изменение термодинамического потенциала системы. Отношение бесконечно малого изменения величины свободной энергии системы к бесконечно малому количеству компонента, внесенному в систему, есть химический потенциал μ i данного компонента в системе:
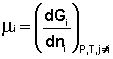 (I.85)
(I.85)
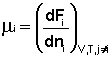 (I.86)
(I.86)
Химический потенциал компонента связан с его парциальным давлением или концентрацией следующими соотношениями:
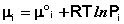 (I.87)
(I.87)
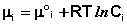 (I.88)
(I.88)
Здесь μ°i – стандартный химический потенциал компонента (Pi = 1 атм., Сi = 1 моль/л.). Очевидно, что изменение свободной энергии системы можно связать с изменением состава системы следующим образом:
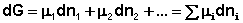 (I.89)
(I.89)
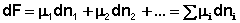 (I.90)
(I.90)
Поскольку условием равновесия является минимум свободной энергии системы (dG = 0, dF = 0), можно записать:
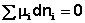 (I.91)
(I.91)
В закрытой системе изменение числа молей одного компонента сопровождается эквивалентным изменением числа молей остальных компонентов; т.е., для приведенной выше химической реакции имеет место соотношение:
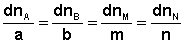 (I.92)
(I.92)
Отсюда можно получить следующее условие химического равновесия в закрытой системе:
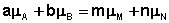 (I.93)
(I.93)
В общем виде условие химического равновесия можно записать следующим образом:
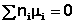 (I.94)
(I.94)
Выражение (I.94) носит название уравнения Гиббса – Дюгема. Подставив в него зависимость химического потенциала от концентрации, получаем:
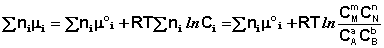 (I.95)
(I.95)
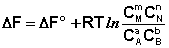 (I.96)
(I.96)
Для изобарно-изотермического процесса аналогичным образом можно получить:
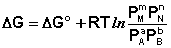 (I.97)
(I.97)
Полученные нами выражения I.96 – I.97 есть изотерма химической реакции . Если система находится в состоянии химического равновесия, то изменение термодинамического потенциала равно нулю; получаем:
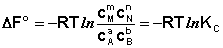 (I.98)
(I.98)
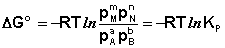 (I.99)
(I.99)
Здесь сi и рi – равновесные концентрации и парциальные давления исходных веществ и продуктов реакции (в отличие от неравновесных Сi и Рi в уравнениях I.96 – I.97).
Поскольку для каждой химической реакции стандартное изменение термодинамического потенциала ΔF° и ΔG° есть строго определенная величина, то произведение равновесных парциальных давлений (концентраций), возведенных в степень, равную стехиометрическому коэффициенту при данном веществе в уравнении химической реакции (стехиометрические коэффициенты при исходных веществах принято считать отрицательными) есть некоторая константа, называемая константой равновесия. Уравнения (I.98, I.99) показывают связь константы равновесия со стандартным изменением свободной энергии в ходе реакции. Уравнение изотермы химической реакции связывает величины реальных концентраций (давлений) реагентов в системе, стандартного изменения термодинамического потенциала в ходе реакции и изменения термодинамического потенциала при переходе из данного состояния системы в равновесное. Знак ΔG (ΔF) определяет возможность самопроизвольного протекания процесса в системе. При этом ΔG° (ΔF°) равно изменению свободной энергии системы при переходе из стандартного состояния (Pi = 1 атм., Сi = 1 моль/л) в равновесное. Уравнение изотермы химической реакции позволяет рассчитать величину ΔG (ΔF) при переходе из любого состояния системы в равновесное, т.е. ответить на вопрос, будет ли химическая реакция протекать самопроизвольно при данных концентрациях Сi (давлениях Рi) реагентов:
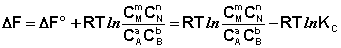 (I.100)
(I.100)
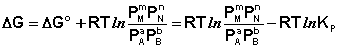 (I.101)
(I.101)
Если изменение термодинамического потенциала меньше нуля, процесс в данных условиях будет протекать самопроизвольно.
1.7.1 Влияние внешних условий на химическое равновесие
При постоянстве внешних условий система может находиться в состоянии равновесия сколь угодно долго. Если изменить эти условия (т.е. оказать на систему какое-либо внешнее воздействие), равновесие нарушается; в системе возникает самопроизвольный процесс, который продолжается до тех пор, пока система опять не достигнет состояния равновесия (уже при новых условиях). Рассмотрим, как влияют на положение равновесия некоторые факторы.
1.7.2 Влияние давления и концентрации
Рассмотрим несколько возможных случаев смещения равновесия.
1. В систему добавлено исходное вещество. В этом случае
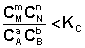 ;
; 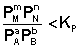 ;
;
По уравнению изотермы химической реакции (I.100 – I.101) получаем: ΔF 0; ΔG > 0. Химическое равновесие будет смещено влево (в сторону расходования продуктов реакции и образования исходных веществ).
3. Изменено общее давление (для реакций в газовой фазе).
Парциальные давления всех компонентов Рi в этом случае изменяются в одинаковой степени; направление смещения равновесия будет определяться суммой стехиометрических коэффициентов Δn.
Учитывая, что парциальное давление газа в смеси равно общему давлению, умноженному на мольную долю компонента в смеси (Рi = РХi), изотерму реакции можно переписать в следующем виде (здесь Δn = Σ(ni) прод – Σ(ni) исх):
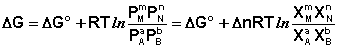 (I.102)
(I.102)
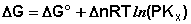 (I.103)
(I.103)
Примем, что Р2 > Р1. В этом случае, если Δn > 0 (реакция идет с увеличением числа молей газообразных веществ), то ΔG > 0; равновесие смещается влево. Если реакция идет с уменьшением числа молей газообразных веществ (Δn изобару Вант-Гоффа :
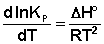 (I.06)
(I.06)
Рассуждая аналогичным образом, для процесса, проходящего в изохорных условиях, можно получить изохору Вант-Гоффа :
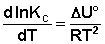 (I.107)
(I.107)
Изобара и изохора Вант-Гоффа связывают изменение константы химического равновесия с тепловым эффектом реакции в изобарных и изохорных условиях соответственно. Очевидно, что чем больше по абсолютной величине тепловой эффект химической реакции, тем сильнее влияет температура на величину константы равновесия. Если реакция не сопровождается тепловым эффектом, то константа равновесия не зависит от температуры.
Экзотермические реакции: ΔH° 0 (ΔU° > 0). В этом случае температурный коэффициент логарифма константы равновесия положителен; повышение температуры увеличивает величину константы равновесия (смещает равновесие вправо).
Графики зависимостей константы равновесия от температуры для экзотермических и эндотермических реакций приведены на рис. I.4.
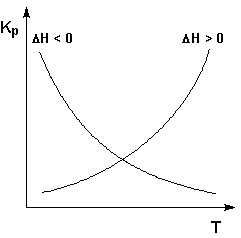
Рис. 1.4 Зависимость константы равновесия от температуры.
Действие рассмотренных нами факторов (давления, концентрации и температуры), равно как и любых других, на систему, находящуюся в состоянии равновесия, обобщает принцип смещения равновесия , называемый также принципом Ле Шателье – Брауна :
Если на систему, находящуюся в состоянии истинного равновесия, оказывается внешнее воздействие, то в системе возникает самопроизвольный процесс, компенсирующий данное воздействие.
Принцип Ле Шателье – Брауна является одним из следствий второго начала термодинамики и применим к любым макроскопическим системам, находящимся в состоянии истинного равновесия.
Copyright © С. И. Левченков, 1996 — 2005.
ВАНТ-ГОФФА ПРАВИЛО
ВАНТ-ГОФФА ПРАВИЛО. Почти все химические реакции при повышении температуры идут быстрее. Зависимость скорости реакции от температуры описывается уравнением Аррениуса:
k = Ae –E a/RT, где k – константа скорости реакции, А – не зависящая от температуры константа (ее называют предэкспоненциальным множителем), Еа– энергия активации, R – газовая постоянная, Т – абсолютная температура. В школьных учебниках зависимость скорости реакции от температуры определяют в соответствии с так называемым «правилом Вант-Гоффа», которое в 19 в. сформулировал голландский химик Якоб Вант-Гофф. Это чисто эмпирическое правило, т.е. правило, основанное не на теории, а выведенное из опытных данных. В соответствии с этим правилом, повышение температуры на 10° приводит к увеличению скорости в 2–4 раза. Математически эту зависимость можно выразить уравнением v2v1 = g (T2 – T 1 )/10 , где v1 и v2– скорости реакции при температурах Т1 и Т2; величина g называется температурным коэффициентом реакции. Например, если g = 2, то при Т2– Т1 = 50 о v2/v1 = 2 5 = 32, т.е. реакция ускорилась в 32 раза, причем это ускорение никак не зависит от абсолютных величин Т1 и Т2, а только от их разности.
Однако из уравнения Аррениуса следует, что температурный коэффициент реакции зависит как от энергии активации, так и от абсолютной температуры. Для данной реакции с определенным значением Еа ускорение при повышении температуры на 10° будет тем больше, чем ниже температура. Это почти очевидно и без расчетов: повышение температуры от 0 до 10° С должно сказаться на скорости реакции значительно сильнее, чем такое же повышение температуры, например, от 500 до 510° С.
С другой стороны, для данного температурного интервала ускорение реакции будет тем сильнее, чем больше ее энергия активации. Так, если энергия активации реакции мала, то такая реакция идет очень быстро, и при повышении температуры на 10° С ее скорость почти не изменяется. Для таких реакций температурный коэффициент намного меньше 2. Для реакций же с большой энергией активации, которые при невысоких температурах идут медленно, ускорение при повышении температуры на 10° С может значительно превысить 4-кратное.
Например, реакция диоксида углерода со щелочным раствором с образованием гидрокарбонат-иона (СО2 + ОН® НСО3 – ) имеет энергию активации 38,2 кДж/моль, поэтому при повышении температуры, например, от 50 до 60° С эта реакция ускорится всего в 1,5 раза. В то же время реакция распада этилбромида на этилен и бромоводород (С2Н5Вr ® С2Н4 + НВr) с энергией активации 218 кДж/моль ускорится при повышении температуры от 100 до 110 o С в 6,3 раза (правда, в этом интервале температур реакция идет очень медленно). Кинетика реакции атомов водорода с этаном H + C2H6 ® H2 + C2H5 была изучены в широком температурном интервале – от 300 до 1100 К (27–827° С). Для этой реакции Eа = 40,6 кДж/моль. Следовательно, повышение температуры на 10° вызовет увеличение скорости реакции в 1,7 раза в интервале 300–310 K и только в 1,04 раза в интервале 1090–1100 K. Так что при высоких температурах скорость этой реакции практически не зависит от температуры. А для реакции присоединения атома водорода к двойной связи H + C2H4 ® C2H5 энергия активации мала (Eа = 3,4 кДж/моль, так что ее скорость слабо зависит от температуры в широком температурном интервале. И только при температурах намного ниже 0° С начинает сказываться наличие активационного барьера.
Подобных примеров можно привести множество. Очевидно, что правило Вант-Гоффа противоречит не только уравнению Аррениуса, но и многим экспериментальным данным. Откуда же оно взялось и почему нередко выполняется?
Если в приведенном выше математическом выражении для правила Вант-Гоффа подставить вместо скоростей v1 и v2 для данной реакции их зависимости от температуры, в соответствии с уравнением Аррениуса, то после сокращения предэкспоненциальных множителей получим следующее выражение: g = vT +10/vT = е –Е а/R(Т+10)/е –Е а/RТ = е (Еа/R)[1/Т – 1/(T+10)] . Логарифмироване этого уравнения дает: lng = (Eа/R)[1/T – 1/(T + 10)], откуда Еа = Rlng T(T + 10)/10 = 0,83lngT(T + 10). Энергия активации не является функцией температуры, эта зависимость нужна лишь для удобства последующего анализа. Последнее уравнение – это уравнение параболы, в котором физический смысл имеют только положительные значения. Соответствующая диаграмма ограничена двумя ветвями параболы: при g = 2 получаем Еа = 0,58Т(Т + 10), при g = 4 получаем Еа = 1,16Т(Т + 10). К тем же формулам приходим и при использовании десятичных логарифмов. Соответствующие графики двух парабол, для значений g 2 и 4, приведены на рисунке. Их физический смысл заключается в том, что области выполнения правила Вант-Гоффа соответствует только область между параболами. Таким образом, существуют только определенные соотношения между энергией активации реакции и температурой ее проведения, при которых правило Вант-Гоффа выполняется. Ниже нижней ветви температурный коэффициент g 4.
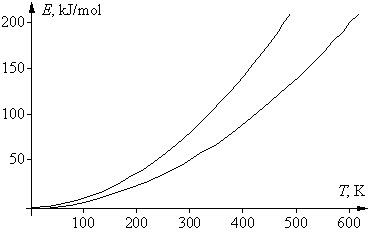
Если посмотреть, какие реакции «укладываются» в указанную довольно узкую область, то окажется, что все эти реакции идут не слишком быстро и не слишком медленно, а с удобной для измерения (при данной температуре) скоростью. Скорость только таких реакций и могли изучать химики во времена Вант-Гоффа. Например, если энергия активации была невелика (меньше 50 кДж/моль), то такая реакция при комнатной температуре заканчивалась за 1–2 секунды; поэтому для изучения ее кинетики следовало значительно понизить температуру, чтобы реакция проходила не быстрее, чем за 10–20 минут. Только в этом случае химики 19 в. успевали отбирать пробы по ходу реакции и анализировать изменение в них концентрации реагентов. Других способов изучения скорости реакции в то время не было. Если это не удавалось (например, водный раствор замерзал), то скорость такой реакции не изучали. Если же энергия активации реакции была велика и при комнатной температуре она шла слишком медленно (многие сутки, или даже недели), то температуру повышали, чтобы реакция шла с удобной для измерения скоростью. И здесь были свои ограничения – например, раствор мог закипеть, т.е. в любом случае исследователи фактически «подстраивали» изучаемую реакцию под область между двумя параболами.
Сейчас химики имеют возможность с помощью различных приборов экспериментально изучать и очень быстрые (идущие в микросекундной области), и очень медленные реакции, для которых температурный коэффициент может быть значительно меньше 2 или значительно больше 4. Поэтому правило Вант-Гоффа, которое, в отличие от уравнения Аррениуса, не имеет четкого физического смысла, представляет лишь чисто исторический интерес и в современной науке не используется. В подавляющем большинстве учебников и монографий по химической кинетике, а также в 5-томной Химической Энциклопедии это правило даже не упоминается. И, тем не менее, если изучаемая реакция идет с удобной для измерения скоростью, например, заканчивается за 30–40 мин, а энергия активации ее еще не измерена, то для предварительной грубой оценки зависимости скорости такой реакции от температуры можно использовать правило Вант-Гоффа. Поэтому это правило приводится во всех школьных учебниках химии.
http://physchem.chimfak.sfedu.ru/Source/PCC/Termodyn_7.htm
http://www.krugosvet.ru/enc/nauka_i_tehnika/himiya/VANT-GOFFA_PRAVILO.html