ВАНТ-ГОФФА ПРАВИЛО
ВАНТ-ГОФФА ПРАВИЛО. Почти все химические реакции при повышении температуры идут быстрее. Зависимость скорости реакции от температуры описывается уравнением Аррениуса:
k = Ae –E a/RT, где k – константа скорости реакции, А – не зависящая от температуры константа (ее называют предэкспоненциальным множителем), Еа– энергия активации, R – газовая постоянная, Т – абсолютная температура. В школьных учебниках зависимость скорости реакции от температуры определяют в соответствии с так называемым «правилом Вант-Гоффа», которое в 19 в. сформулировал голландский химик Якоб Вант-Гофф. Это чисто эмпирическое правило, т.е. правило, основанное не на теории, а выведенное из опытных данных. В соответствии с этим правилом, повышение температуры на 10° приводит к увеличению скорости в 2–4 раза. Математически эту зависимость можно выразить уравнением v2v1 = g (T2 – T 1 )/10 , где v1 и v2– скорости реакции при температурах Т1 и Т2; величина g называется температурным коэффициентом реакции. Например, если g = 2, то при Т2– Т1 = 50 о v2/v1 = 2 5 = 32, т.е. реакция ускорилась в 32 раза, причем это ускорение никак не зависит от абсолютных величин Т1 и Т2, а только от их разности.
Однако из уравнения Аррениуса следует, что температурный коэффициент реакции зависит как от энергии активации, так и от абсолютной температуры. Для данной реакции с определенным значением Еа ускорение при повышении температуры на 10° будет тем больше, чем ниже температура. Это почти очевидно и без расчетов: повышение температуры от 0 до 10° С должно сказаться на скорости реакции значительно сильнее, чем такое же повышение температуры, например, от 500 до 510° С.
С другой стороны, для данного температурного интервала ускорение реакции будет тем сильнее, чем больше ее энергия активации. Так, если энергия активации реакции мала, то такая реакция идет очень быстро, и при повышении температуры на 10° С ее скорость почти не изменяется. Для таких реакций температурный коэффициент намного меньше 2. Для реакций же с большой энергией активации, которые при невысоких температурах идут медленно, ускорение при повышении температуры на 10° С может значительно превысить 4-кратное.
Например, реакция диоксида углерода со щелочным раствором с образованием гидрокарбонат-иона (СО2 + ОН® НСО3 – ) имеет энергию активации 38,2 кДж/моль, поэтому при повышении температуры, например, от 50 до 60° С эта реакция ускорится всего в 1,5 раза. В то же время реакция распада этилбромида на этилен и бромоводород (С2Н5Вr ® С2Н4 + НВr) с энергией активации 218 кДж/моль ускорится при повышении температуры от 100 до 110 o С в 6,3 раза (правда, в этом интервале температур реакция идет очень медленно). Кинетика реакции атомов водорода с этаном H + C2H6 ® H2 + C2H5 была изучены в широком температурном интервале – от 300 до 1100 К (27–827° С). Для этой реакции Eа = 40,6 кДж/моль. Следовательно, повышение температуры на 10° вызовет увеличение скорости реакции в 1,7 раза в интервале 300–310 K и только в 1,04 раза в интервале 1090–1100 K. Так что при высоких температурах скорость этой реакции практически не зависит от температуры. А для реакции присоединения атома водорода к двойной связи H + C2H4 ® C2H5 энергия активации мала (Eа = 3,4 кДж/моль, так что ее скорость слабо зависит от температуры в широком температурном интервале. И только при температурах намного ниже 0° С начинает сказываться наличие активационного барьера.
Подобных примеров можно привести множество. Очевидно, что правило Вант-Гоффа противоречит не только уравнению Аррениуса, но и многим экспериментальным данным. Откуда же оно взялось и почему нередко выполняется?
Если в приведенном выше математическом выражении для правила Вант-Гоффа подставить вместо скоростей v1 и v2 для данной реакции их зависимости от температуры, в соответствии с уравнением Аррениуса, то после сокращения предэкспоненциальных множителей получим следующее выражение: g = vT +10/vT = е –Е а/R(Т+10)/е –Е а/RТ = е (Еа/R)[1/Т – 1/(T+10)] . Логарифмироване этого уравнения дает: lng = (Eа/R)[1/T – 1/(T + 10)], откуда Еа = Rlng T(T + 10)/10 = 0,83lngT(T + 10). Энергия активации не является функцией температуры, эта зависимость нужна лишь для удобства последующего анализа. Последнее уравнение – это уравнение параболы, в котором физический смысл имеют только положительные значения. Соответствующая диаграмма ограничена двумя ветвями параболы: при g = 2 получаем Еа = 0,58Т(Т + 10), при g = 4 получаем Еа = 1,16Т(Т + 10). К тем же формулам приходим и при использовании десятичных логарифмов. Соответствующие графики двух парабол, для значений g 2 и 4, приведены на рисунке. Их физический смысл заключается в том, что области выполнения правила Вант-Гоффа соответствует только область между параболами. Таким образом, существуют только определенные соотношения между энергией активации реакции и температурой ее проведения, при которых правило Вант-Гоффа выполняется. Ниже нижней ветви температурный коэффициент g 4.
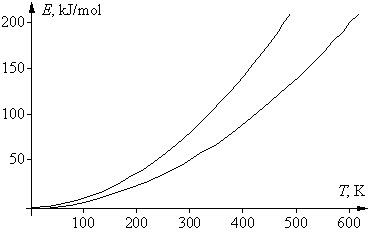
Если посмотреть, какие реакции «укладываются» в указанную довольно узкую область, то окажется, что все эти реакции идут не слишком быстро и не слишком медленно, а с удобной для измерения (при данной температуре) скоростью. Скорость только таких реакций и могли изучать химики во времена Вант-Гоффа. Например, если энергия активации была невелика (меньше 50 кДж/моль), то такая реакция при комнатной температуре заканчивалась за 1–2 секунды; поэтому для изучения ее кинетики следовало значительно понизить температуру, чтобы реакция проходила не быстрее, чем за 10–20 минут. Только в этом случае химики 19 в. успевали отбирать пробы по ходу реакции и анализировать изменение в них концентрации реагентов. Других способов изучения скорости реакции в то время не было. Если это не удавалось (например, водный раствор замерзал), то скорость такой реакции не изучали. Если же энергия активации реакции была велика и при комнатной температуре она шла слишком медленно (многие сутки, или даже недели), то температуру повышали, чтобы реакция шла с удобной для измерения скоростью. И здесь были свои ограничения – например, раствор мог закипеть, т.е. в любом случае исследователи фактически «подстраивали» изучаемую реакцию под область между двумя параболами.
Сейчас химики имеют возможность с помощью различных приборов экспериментально изучать и очень быстрые (идущие в микросекундной области), и очень медленные реакции, для которых температурный коэффициент может быть значительно меньше 2 или значительно больше 4. Поэтому правило Вант-Гоффа, которое, в отличие от уравнения Аррениуса, не имеет четкого физического смысла, представляет лишь чисто исторический интерес и в современной науке не используется. В подавляющем большинстве учебников и монографий по химической кинетике, а также в 5-томной Химической Энциклопедии это правило даже не упоминается. И, тем не менее, если изучаемая реакция идет с удобной для измерения скоростью, например, заканчивается за 30–40 мин, а энергия активации ее еще не измерена, то для предварительной грубой оценки зависимости скорости такой реакции от температуры можно использовать правило Вант-Гоффа. Поэтому это правило приводится во всех школьных учебниках химии.
Растворы. Законы Рауля, Вант-Гоффа
1. Основные понятия и определения.
2. Идеальные растворы. Физико-химические свойства разбавленных растворов неэлектролитов и электролитов. Законы Рауля, Вант-Гоффа.
3. Электролитическая диссоциация. Слабые и сильные электролиты. Степень и константа диссоциации. Активность.
4. Электролитическая диссоциация воды. Водородный показатель. Ионное произведение воды.
5. Гидролиз солей.
Химически чистое вещество представляет собой предельное состояние, которое практически не достигается. Даже особо чистые металлы, получаемые методами вакуумной или зонной плавки, содержат определенное количество примесей, около 10 –6 % (масс.) и по существу являются твердыми растворами. Образование растворов — систем, не подчиняющихся стехиометрическим законам (сохранения массы, постоянства состава, кратных и объемных отношений, эквивалентов), и обладающих переменным составом, существенно изменяет условия протекания химических реакций между компонентами. Растворы широко распространены в природе (особенно водные) и играют важную роль во многих отраслях промышленности и техники.
Изучение физико–химических свойств растворов, условий их образования обогащает и углубляет представления о механизме и закономерностях протекания химических процессов.
ВОПРОС 1. Раствор — гомогенная физико–химическая система переменного состава, состоящая из двух или более компонентов и продуктов их взаимодействия.
По агрегатному состоянию растворы разделяют на жидкие, твердые и газообразные (рис.1). Наиболее значительную роль в химии и особенно в биохимии играют жидкие растворы.
Компонентами раствора являются растворитель (среда) и растворенное вещество, равномерно распределенное в растворителе в виде молекул или ионов. Под растворителем обычно понимают то вещество, которое находится в таком же агрегатном состоянии, как и раствор в целом. Если вещества, составляющие раствор, имеют одинаковое агрегатное состояние (например, спирт и вода), то растворителем называют то из веществ, которое имеется в большем количестве.
Относительные количества компонентов раствора можно изменять в довольно широких пределах.

Рис.1. Классификация растворов
Процесс растворения вещества нельзя считать чисто физическим явлением. При растворении происходит химическое взаимодействие частиц растворенного вещества с молекулами растворителя, в результате чего образуются соединения, называемые сольватами. Если растворителем является вода, то эти соединения называют гидратами.
Как правило, гидраты — соединения менее прочные, чем обычные химические соединения. Однако часто гидратная вода настолько прочно связывается с молекулами растворенного вещества, что при кристаллизации входит в состав твердой фазы. Эту воду называют кристаллизационной водой, а сами кристаллические образования — кристаллогидратами, состав которых выражают формулами CuSO4·5H2O, FeSO4·7H2O, Na2SO4·10H2O и т.п.
Процесс растворения кристаллических веществ в воде состоит из двух последовательных стадий, каждая из которых сопровождается тепловым эффектом:
1 стадия — разрушение кристаллической решетки растворяемого вещества на отдельные частицы — идет с поглощением теплоты (?Н1 > 0).
2 стадия — взаимодействие частиц растворенного вещества с молекулами воды (гидратация) — идет с выделением теплоты (?Н2
Таким образом, тепловой эффект растворения ?Н является алгебраической суммой двух тепловых эффектов: ?Н = ?Н1 + ?Н2.
Растворимостью называют способность данного вещества растворяться в том или ином растворителе. Количественно растворимость характеризуется массой вещества или количеством вещества, которое может раствориться при данной температуре в определенном количестве растворителя.
Раствор, в котором при данной температуре вещество больше растворяться не может, называют насыщенным.
Если вещество еще может растворяться в данном растворе, то оно называется ненасыщенным.
Из сказанного следует, что концентрация вещества в насыщенном растворе равна его растворимости, концентрация ненасыщенного раствора всегда меньше величины растворимости.
Растворимость веществ обычно зависит от температуры — самостоятельное изучение.
Часто растворы подразделяют на разбавленные и концентрированные.— самостоятельное изучение.
2. Коллигативные свойства растворов.
Коллигативными называют такие свойства раствора, которые зависят только от концентрации растворенного вещества и природы растворителя, но не зависят от природы растворенного вещества.
К этим свойствам относятся: 1) понижение давления насыщенного пара над раствором; 2) понижение температуры замерзания; 3) повышение температуры кипения; 4)осмотическое давление. Далее мы будем рассматривать их в применении к растворам нелетучих веществ, т.е. давление пара которых над раствором пренебрежимо мало.
Коллигативные свойства раствора — это свойства идеального раствора.
Идеальным называют раствор, в котором не происходят химические реакции между компонентами, а сила межмолекулярного взаимодействия однородных и разнородных частиц одинакова.
Соответственно, образование этих растворов не сопровождается тепловым эффектом (?Н = 0) и каждый компонент ведет себя в растворе независимо от других компонентов. К идеальным растворам по своим свойствам приближаются лишь очень разбавленные растворы, т.е. растворы с очень низкой концентрацией растворенного вещества.
Понижение давления насыщенного пара над раствором.
Одно из важнейших явлений, характерных для жидких растворов, — понижение давления насыщенного пара растворителя над раствором по сравнению с давлением насыщенного пара чистого растворителя.
Давление насыщенного пара при данной температуре характеризует равновесие между жидким и газообразным состоянием вещества в закрытой системе, возникающее на границе раздела фаз вследствие выравнивания скоростей эндотермического процесса испарения и экзотермического процесса конденсации. В растворе концентрация молекул растворителя в поверхностном слое меньше, чем в чистом растворителе (молекулы растворенного вещества сольватируясь (гидратируясь) занимают часть поверхности раствора), поэтому равновесие достигается при меньшем давлении насыщенного пара.
Выражение Р 0 А–РА = ?РА = Р 0 А· Nв
количественно выражает связь между концентрацией растворенного вещества в растворе и давлением насыщенного пара растворителя над раствором. Его называют законом Рауля:понижение давления насыщенного пара растворителя А над раствором ?РАпропорционально мольной доле растворенного нелетучего вещества ·Nв.
Здесь Р 0 А и РА — давления насыщенного пара растворителя соответственно над чистым растворителем и над раствором; ?РА – разность между давлениями насыщенного пара растворителя над раствором РА и растворителем Р 0 А.
Из выражения закона Рауля следует, что с увеличением содержания нелетучего растворенного компонента давление пара растворителя над раствором уменьшается.
Из закона Рауля возникают два следствия. Согласно одному из них температура кипения раствора выше температуры кипения растворителя. Это обусловлено тем, что давление насыщенного пара растворителя над раствором становится равным атмосферному давлению (условие кипения жидкости) при более высокой температуре, чем в случае чистого растворителя.
Повышение температуры кипения ?Ткип. пропорционально моляльности раствора.
?Ткип. = Кэ ·Сm ,
где Кэ — эбуллиоскопическая постоянная (ebullir — выкипать, skopeo— смотрю). Она означает повышение температуры кипения раствора при его моляльной концентрации, равной 1.
Вспомнив определение моляльности раствора можно записать:
?Ткип. = Кэ mр.в.·1000/М·mр-ля
Согласно второму следствию из закона Рауля температура замерзания (кристаллизации) раствора ниже температуры замерзания (кристаллизации) чистого растворителя. Раствор замерзает при более низкой температуре, чем растворитель, так как молекулы растворенного вещества при понижении температуры препятствуют формированию кристаллической решетки при затвердевании раствора.
Понижение температуры замерзания (кристаллизации) ?Тзам. пропорционально моляльности раствора:
или ?Тзам. = Кк mр.в.·1000/М·mр-ля
где Кк — криоскопическая постоянная (kryos — лед, холод). Она означает понижение температуры затвердевания раствора при его моляльной концентрации, равной 1.
Значения Кэ и Кк зависят от природы растворителя и не зависят от природы растворенного вещества и его концентрации.
Значения Кэ и Кк — справочные величины.
Осмотическое давление —
3. Электролитическая диссоциация.
Итак, мы с вами познакомились с основными свойствами разбавленных растворов: понижением давления насыщенного пара над раствором, повышением температуры кипения и понижением температуры замерзания растворов и осмотическим давлением.
Что можно еще добавить к этому вопросу? А добавить нужно следующее: исследование свойств разбавленных растворов многочисленных веществ показало, что все вещества могут быть разделены на две группы.
К первой группе принадлежат те вещества, разбавленные растворы которых оказывают осмотическое давление, равное вычисленному по закону Вант–Гоффа. К таким веществам относятся сахароза, глюкоза, глицерин и многие другие вещества–неэлектролиты. Неэлектролиты — вещества, не распадающиеся в растворах на ионы, и соответственно, не проводящие электрический ток.
Вторую группу составляют соли, основания и кислоты. Эти вещества называют электролитами(электролиты — вещества, распадающиеся в растворах на ионы, и проводящие электрический ток). Осмотическое давление разбавленных растворов этих веществ оказалось больше, чем можно было бы ожидать по теории Вант–Гоффа, причем настолько больше, что это нельзя было объяснить погрешностями опытов. Также и другие свойства растворов этих веществ оказались значительно большими, чем для веществ первой группы.
Желая примирить эти кажущиеся противоречия, Ватг–Гофф ввел в свое уравнение коэффициент i, в дальнейшем получивший название изотонического коэффициента.
Вант–Гофф установил, что коэффициент i зависит от природы растворенного вещества, а иногда и от концентрации раствора, но каков физический смысл этого коэффициента он не знал. Далее было установлено, что для расчета давления насыщенного пара над раствором, повышения температуры кипения или понижения температуры замерзания растворов электролитов в соответствующие уравнения вводиться коэффициент i:
?Ткип. = i·Кэ ·Сm
?Тзам. = i· Кк ·Сm
Но в чем же все-таки физический смысл изотонического коэффициента?
Ознакомившись с работой Вант–Гоффа, Аррениус пришел к выводу, что коэффициент i правильнее было бы назвать не изотоническим, а коэффициентом диссоциации, учитывающим увеличение числа частиц в растворе в результате электролитической диссоциации.
Электролитической диссоциацией называют частичный или полный распад растворенного вещества на ионы в результате взаимодействия с растворителем.
По мнению Аррениуса, диссоциация электролитов на ионы приводит к значительному увеличению общего числа частиц в растворе, по сравнению с числом частиц в растворах неэлектролитов. А поскольку осмотическое давление, давление насыщенного пара растворителя над раствором, температуры кипения и замерзания растворов (т.е. все коллигативные свойства растворов) зависят от числа частиц растворенного вещества, находящихся в единице объема раствора, то с увеличением этого числа эти свойства тоже увеличиваются.
Таким образом, изотонический коэффициент Вант–Гоффа является мерой увеличения частиц в растворе вследствие электролитической диссоциации.
или иными словами, изотонический коэффициент — величина, показывающая во сколько раз значения ?РА, ?, ?Ткип. и ?Тзам. в растворах электролитов больше тех же величин, в растворах неэлектролитов.
Если бы электролиты полностью диссоциировали на ионы, то коллигативные свойства растворов электролитов всегда были бы в целое число раз больше значений, наблюдаемых в растворах неэлектролитов. Но еще Вант–Гофф установил, что коэффициент i выражается дробными числами, которые с разбавлением раствора возрастают, приближаясь к целым числам.
Аррениус объяснил это факт тем, что лишь часть электролита диссоциирует в растворе на ионы, и ввел понятие степени диссоциации. Степенью диссоциации электролита (?) называется отношение числа его молекул, распавшихся в данном растворе на ионы, к общему числу его молекул в растворе. Т.е. ? =
Позже было установлено, что электролиты можно разделить на две группы: сильные и слабыеэлектролиты.
Сильные электролиты в водных растворах диссоциированы практически нацело. (?
1, а по-честному ? >0,3). Понятие степени диссоциации к ним практически не применимо, а отклонение изотонического коэффициента i от целочисленных значений объясняется другими причинами.
Слабые электролиты в водных растворах диссоциируют только частично, и в растворе устанавливается динамическое равновесие между недиссоциированными молекулами и ионами.
К сильным электролитам относятся:
1) почти все соли (кроме CdCl2, HgCl2 и некоторых других);
2) большинство минеральных кислот (например, H2SO4, HNO3, HCl, HBr, HI, HClO4);
3) основания щелочных и щелочноземельных металлов.
К слабым электролитам относятся:
1) почти все органические кислоты;
3) гидроксиды почти всех металлов (за исключением щелочных и щелочно–земельных металлов), а также гидроксид аммиака.
К слабым электролитам относится также вода.
К равновесию, которое устанавливается в растворе слабого электролита между молекулами и ионами, можно применить законы химического равновесия. Например, для диссоциации уксусной кислоты.
По этой ссылке вы найдёте полный курс лекций по математике:
Пусть температура, при которой протекает обратимая реакции электролитической диссоциации, постоянна, тогда в данной системе наступит равновесие, и концентрации непродиссоциировавших молекул СН3СООН, а также ионов – продуктов диссоциации, становятся постоянными. Получаем:
Константа равновесия в данном случае называется константой диссоциации, т.к. она характеризует способность электролита диссоциировать на ионы: чем больше КД, тем больше ионов в растворе.
КД зависит от природы электролита, природы растворителя и, как любая константа равновесия, от температуры, но не зависит от концентрации электролита.
Многоосновные кислоты диссоциируют ступенчато. Так, диссоциация Н3РО4 можно представить уравнениями:
Всегда К1>К2>К3, поэтому в растворах таких кислот в заметных количествах присутствуют только те ионы, которые образуются только по первой ступени диссоциации.
Понятие константы диссоциации неприменимо к сильным электролитам, т.к. в растворах сильных электролитов отсутствует равновесие между молекулами и ионами (молекулы практически полностью распадаются на ионы).
При добавлении одного из продуктов диссоциации, т.е. одноименного иона, будет смещать равновесие влево. Следовательно, введение в раствор слабого электролита одноименных ионов подавляет диссоциацию электролита, а значит и уменьшает степень диссоциации.
Определим зависимость между константой диссоциации, степенью диссоциации и концентрацией раствора.
Пусть С – первоначальная концентрация уксусной кислоты, моль/л;
— степень ее диссоциации, тогда С — концентрация продиссоциированных молекул кислоты; С-С = С(1- )- концентрация недиссоциированных молекул кислоты. Поскольку из 1 молекулы СН3СООН образуется 1 ион Н + и 1 ион СН3СОО — , то [Н + ]=[ СН3СОО — ]= С , подставляем это в уравнение константы диссоциации:
Для слабого электролита при не слишком низкой концентрации степень диссоциации очень мала, и 1-
Эти выражения характеризуют закон разбавления Оствальда: степень диссоциации слабого электролита обратно пропорциональна корню квадратному из концентрации раствора, т.е. увеличивается с разбавлением раствора.
В данной форме закон применим для бинарных электролитов.
4. Электролитическая диссоциация воды. Водородный показатель.
Особо следует остановиться на равновесии электролитической диссоциации наиболее распространенного природного растворителя — воды.
Вода, представляет собой слабый электролит. В ничтожно малой степени молекулы воды диссоциируют в соответствии с уравнением:
Этот процесс называется самоионизацией или автопротолизом. В данном случае мы записали упрощенную схему разложения воды.
С учетом гидратации ионов водорода уравнение диссоциации можно представить так:
Хотя установлено, что, кроме иона гидроксония Н3О + , в растворе существуют и более сложные гидраты: диаквапротон Н5О2 + , триаквапротон Н7О3 + и др.
Константу диссоциации воды можно записать в виде:
При 25°С (298 К) константа диссоциации воды равна 2·10 —16 . В знаменателе стоит молярная концентрация недиссоциированных молекул воды. Поскольку степень диссоциации воды очень мала, то концентрацию недиссоциированных молекул можно считать равной общей концентрации молекул воды, которая постоянна при Т=const. Поэтому константа равновесия процесса диссоциации воды определяется произведением равновесных молекулярных концентраций ионов:
Общая концентрация молекул воды можно рассчитать, разделив массу 1 литра воды на массу ее моля:
1000/18 = 55 моль/л
Подставляя эту величину в последнее уравнение, получим:
[Н + ]·[ОН – ] = 2·10 –16 · 55 = 10 –14 = Кw
Произведение концентраций ионов [Н + ] и [ОН – ] называют ионным произведением воды Кw.
Диссоциация воды есть эндотермический процесс, поэтому ионное произведение воды Кwувеличивается с ростом температуры.
Как всякая константа равновесия, Кw не зависит от концентраций ионов Н + и ОН – в растворе. Так, если в Н2О добавить кислоты, концентрация ионов Н + резко возрастет. Тогда за счет подавления диссоциации Н2О равновесие этого процесса сместится влево и концентрация ионов ОН – в растворе уменьшится, но ионное произведение Н2О останется неизменным.
Для указания концентрации ионов водорода в растворе используют водородный показатель
а для обозначения концентрации ионов гидроксида — гидроксидный показатель:
Так как в соответствии с уравнением диссоциации воды концентрации ионов H + и ОH – в воде одинаковы, и исходя из выражения ионного произведения воды (при 298 К [Н + ]·[ОН – ] = Кw = 10 –14 ) имеем, что:
[H + ] = [ОH – ] = ?Кw = 10 –7 моль/л
Следовательно pH = pОH = 7. Такие растворы называют нейтральными.
В кислой среде [H + ] > [ОH – ], pH 7;
В щелочных растворах [H + ] – ], pH > 7, а pОH
Сок лимонный рН = 2,1
Сок апельсиновый рН = 2,8
Сок желудочный рН = 1,4
Сухое вино рН = 3,5
Кофе черный рН = 5
Вода дождевая рН = 6,5
Вода дистиллированная рН = 7
Аммиак (бытовой) рН = 11,9
Не следует забывать, что для любой среды pH + pОH = 14.
Итак, при расчете pH слабых электролитов пользуются выражением
Концентрацию ионов водорода в растворе слабых кислот определяют по уравнению Оствальда (Кд= ? 2 ·с) или
[H + ] = ?·с = ?Кд·с (где ? – степень диссоциации, с – концентрация кислоты)
Аналогично определяется концентрация ионов гидроксида:
Возможно вам будут полезны данные страницы:
5. Гидролиз солей
Процесс растворения и сольватации многих солей в воде сопровождается образованием нейтральных растворов.
| Однако некоторые соли взаимодействуют |
с водой так, что образуются кислотные или щелочные растворы. В этом можно убедиться, если добавить немного фенолфталеина в раствор сульфита натрия Na2SO3 или прилить к раствору сульфата алюминия Al2(SO4)3 несколько капель лакмуса. Малиновое окрашивание индикатора в первом растворе и красное во втором убедительно свидетельствует, что растворы не нейтральны. Раствор сульфита натрия имеет щелочную реакцию, а сульфата алюминия — кислую. Но как в этих растворах появились избыточные Н + — и ОН – -ионы? Они появились из воды в результате протекания процесса гидролиза.
Гидролиз солей — это химическая реакция ионного обмена между водой и растворенными в ней солями, приводящее к увеличению кислотности или щелочности раствора.
Рассмотрим почему и как протекает эта реакция:
Все ионы соли в растворе окружены гидратной оболочкой. На ионах сосредоточен значительный электрический заряд, а молекулы воды поляризованы, причем поляризация усиливается за счет электрического поля, индуцируемого зарядом иона (так называемая наведенная поляризация). Наиболее сильно эффект дополнительной поляризации проявляется в молекулах из ближайшего к иону слоя гидратной оболочки. Это делает возможным разрыв О–Н связи в молекуле воды и образование новой связи Kat–OH или An–H (Kat – катион, An – анион). Схематически этот процесс можно представить следующим образом:
Kat n + + НОН ? Kat–OH ( n –1) + H + (а)
An n – + НОН ? An–H ( n –1) + ОН – (б)
Образовавшиеся частицы могут быть электронейтральными (n = 1) или нести заряд, в зависимости от зарядов исходных ионов. Если обе реакции (а) и (б) обратимы, а это возможно только в случае когда Kat–OH и An–Н являются сильными электролитами, т.е. мгновенно подвергаются диссоциации, то в растворе не возникает избыточного количества ни протонов, ни гидроксид ионов. Среда остается нейтральной. В этом случае говорят, что гидролиз не протекает. Если одна из стадий обратима, а другая нет, то в растворе возникнет некоторое избыточное, по сравнению с нейтральным раствором, количество этих ионов. Таким образом, раствор соли уже не будет иметь нейтральную среду, а приобретет кислую или щелочную реакцию. Данный процесс называется гидролизом. И он возможен только в случае, когда либо Kat–OH, либо An–Н являются слабыми электролитами.
Иными словами, для того чтобы в растворе соли протекал процесс гидролиза, необходимо, чтобы соль в своем составе содержала либо катионы слабого основания, либо анионы слабой кислоты, либо и те и другие, которые могут связывать соответственно ионы ОН – и Н + с образованием слабого электролита.
Рассмотрим возможные случаи гидролиза.
1) Для солей, образованных сильной кислотой и сильным основанием:
NaCl — гидролизу не подвергается, рН = 7
2) Для солей, образованных слабой кислотой и сильным основанием (гидролиз соли по аниону):
гидролиз соли по аниону
3) Для солей, образованных слабым основанием и сильной кислотой (гидролиз соли по катиону):
4) Для солей, образованных слабым основанием и слабой кислотой (гидролиз соли и по катиону и по аниону):
В данном случае реакцию среды определяет то соединение, которое имеет большую Кд
В рассмотренных нами случаях гидролиз шел по первой ступени, то есть присоединялся один ион Н + или ОН – . Если валентность атома металла или кислотного остатка больше единицы, то он может присоединять большее количество Н + или ОН – . В таком случае гидролиз идет ступенчато:
Гидролиз по первой ступени называется простым, обратимым. Гидролиз, сопровождающийся уходом продуктов реакции из зоны реакции или образованием осадка, часто называют необратимым (полным, сложным). Со(ОН)2 — осадок, значит рассмотренный нами пример является случаем необратимого гидролиза.
При полном гидролизе реализуются не только первая, но и последующая ступени.
На практике возникает необходимость расчета рН раствора соли после гидролиза, а также степени гидролиза — глубины протекания гидролиза.
Степень гидролиза (h) — отношение числа молей соли, подвергшихся гидролизу, к исходному количеству растворенной соли в молях.
или
Степень гидролиза (h) — отношение концентрации гидролизованных молекул Сгидр. к исходной концентрации растворенных молекул электролита:
h = cгидр. / c
Степень гидролиза, как правило, невелика. Так, в 0,1 М растворе СH3СООNa при 298 К составляет примерно 10 –4 , т.е в этом растворе гидролизована лишь одна из 10000 молекул. Причина столь низкой степени гидролиза кроется в том, что один из участников реакции — вода является очень слабым электролитом. Поэтому положение равновесия реакции гидролиза сильно смещено в сторону исходных веществ. Степень гидролиза возрастает с увеличением температуры, поскольку гидролиз — процесс эндотермический.
Равновесие гидролиза, как любое химическое равновесие, можно охарактеризовать величиной константы равновесия. Эта величина называется константой гидролиза Кг. Выражение константы гидролиза хлорида алюминия по первой ступени имеет вид:
I ступень: Al 3+ + H2O ? AlOH 2+ + H +
К 1 г = [AlOH 2+ ]·[H + ] / [Al 3+ ]
Константа гидролиза и степень гидролиза связаны между собой отношением, аналогичным закону разбавления Оствальда:
Концентрация [ОН – ] = h·с = ?Кг.·с, поэтому при гидролизе по аниону:
Концентрация [Н + ] = h·с = ?Кг.·с, следовательно, при гидролизе по катиону:
Итак, при гидролизе солей, образованных слабыми кислотами или (и) основаниями, происходит подщелачивание или подкисление раствора. Степень гидролиза возрастает с разбавлением раствора и при увеличении температуры.
Гидролиз играет важную роль в природных и технологических процессах. Например, расщепление пищи в желудочно–кишечном тракте идет по реакции гидролиза ее компонентов. Гидролиз используется в технике при получении ценных продуктов из древесины, жиров и других веществ.
Присылайте задания в любое время дня и ночи в ➔ 

Официальный сайт Брильёновой Натальи Валерьевны преподавателя кафедры информатики и электроники Екатеринбургского государственного института.
Все авторские права на размещённые материалы сохранены за правообладателями этих материалов. Любое коммерческое и/или иное использование кроме предварительного ознакомления материалов сайта natalibrilenova.ru запрещено. Публикация и распространение размещённых материалов не преследует за собой коммерческой и/или любой другой выгоды.
Сайт предназначен для облегчения образовательного путешествия студентам очникам и заочникам по вопросам обучения . Наталья Брильёнова не предлагает и не оказывает товары и услуги.
Правило Вант- Гоффа. Уравнение Аррениуса.
для студентов направления 6070104 «Морской и речной транспорт»
«Эксплуатация судового электрооборудования и средств автоматики»,
направления 6.050702 «Электромеханика» специальности
«Электрические системы и комплексы транспортных средств»,
«Электромеханические системы автоматизации и электропривод»
дневной и заочной форм обучения
Тираж_____экз. Подписано к печати_____________.
Заказ №________. Объем 1,08 п.л.
Изд-во “Керченский государственный морской технологический университет”
98309 г. Керчь, Орджоникидзе, 82.
Правило Вант- Гоффа. Уравнение Аррениуса.
Согласно эмпирическому правилу Вант — Гоффа, сформулированному около 1880г., скорость большинства реакций увеличивается в 2-4 раза при повышении температуры на 10 градусов, если реакция проводится при температуре, близкой к комнатной. Например, время полуразложения газообразного оксида азота (V) при 35°С составляет около 85мин., при 45°С-около 22мин. и при 55°С — около 8мин.
Мы уже знаем, что при любой постоянной температуре скорость реакции описывается эмпирическим кинетическим уравнением, представляющим в большинстве случаев (за исключением реакции с весьма сложным механизмом) произведение константы скорости на концентрации реагентов в степенях, равных порядкам реакции. Концентрации реагентов практически не зависят от температуры, порядки, как показывает опыт,- тоже. Следовательно, за резкую зависимость скорости реакции от температуры ответственны константы скоростей. Зависимость константы скорости от температуры принято характеризовать температурным коэффициентом скорости реакции, которыйпредставляет собой отношение констант скорости при температурах, отличающихся на 10 градусов
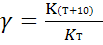 ;
; 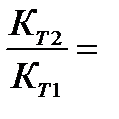
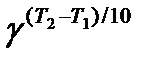 ;
; 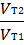 = ;
= ; 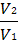 =
= 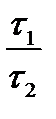
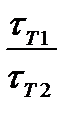 =
=
и который по правилу Вант — Гоффа равен приблизительно 2-4.
Попытаемся объяснить наблюдаемые высокие значения температурных коэффициентов скоростей реакции на примере гомогенной реакции в газовой фазе с позиций молекулярно-кинетической теории газов. Чтобы молекулы взаимодействующих газов прореагировали друг с другом, необходимо их столкновение, при котором одни связи рвутся, а другие образуются, в результате чего и появляется новая молекула — молекула продукта реакции. Следовательно, скорость реакции зависит от числа столкновений молекул реагентов, а число столкновений, в частности, — от скорости хаотического теплового движения молекул. Скорость молекул и соответственно число столкновений растут с температурой. Однако только повышение скорости молекул не объясняет столь быстрого роста скоростей реакций с температурой. Действительно, согласно молекулярно-кинетической теории газов средняя скорость молекул пропорциональна квадратному корню из абсолютной температуры, т.е, при повышении температуры системы на 10 градусов, скажем, от 300 до 310К, средняя скорость молекул возрастет лишь в 310/300 = 1,02 раза — гораздо меньше, чем требует правило Вант -Гоффа.
Таким образом, одним только увеличением числа столкновений нельзя объяснить зависимость констант скоростей реакции от температуры. Очевидно, здесь действует еще какой-то важный фактор. Чтобы вскрыть его, обратимся к более подробному анализу поведения большого числа частиц при различных температурах. До сих пор мы говорили о средней скорости теплового движения молекул и ее изменении с температурой, но если число частиц в системе велико, то по законам статистики отдельные частицы могут иметь скорость и соответственно киетическую энергию, в большей или меньшей степени отклоняющуюся от среднего значения для данной температуры. Эта ситуация изображена на рис. (3.2), который
показывает, как распределены части-
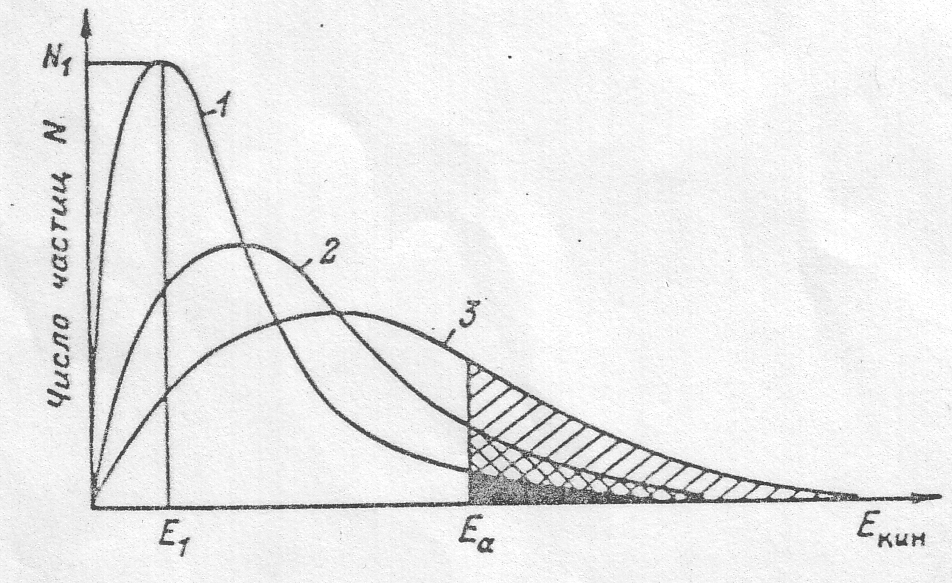
3.2. Распределение частиц по кинетической энергии при различных температурах:
2-Т2; 3-Т3; Ti / — энергия активации химической реакции с участием катализатора.
http://natalibrilenova.ru/rastvoryi-zakonyi-raulya-vant-goffa/
http://helpiks.org/1-49622.html