Зависимость фермента от количества субстрата описывает ферментативная кинетика
Уравнения Михаэлиса-Ментен и Лайнуивера-Берка
Общую теорию ферментативной кинетики и зависимость активности фермента от субстрата.описали Л.Михаэлис и М.Л.Ментен, выразив его в своем уравнении. Бриггс и Холдейн усовершенствовали их уравнение, введя введя в него константу Михаэлиса (Km), определяемую экспериментально.
Уравнение Михаэлиса-Ментен показывает взаимосвязь максимально возможной скорости, реальной скорости реакции, константы Михаэлиса и концентрации субстрата. Так как пользоваться графиком, построенным в прямых координатах V и [S] для точных расчетов неудобно, то Г.Лайнуивер и Д.Бэрк преобразовали уравнение Бриггса–Холдейна в обратные координаты.
Уравнение Михаэлиса-Ментен
Уравнение Лайнуивера-Бэрка
На самом деле уравнение Михаэлиса-Ментен в данном виде предложили Бриггс и Холдейн, но в честь основоположников оно носит название Михаэлиса-Ментен.
Выделяют три основных решения уравнения Михаэлиса-Ментен:
1. Концентрация субстрата равна величине константы Михаэлиса ([S] = Km).
В этом случае, решая уравнение Михаэлиса-Ментен, получаем, что скорость реакции V будет равна половине максимальной Vmax.(V = ½ Vmax).
В математическом смысле Km соответствует концентрации субстрата при которой скорость реакции равна половине максимальной. Ее биологический смысл заключается в характеристике сродства фермента к субстрату, а именно: увеличение величины Кm означает снижение сродства фермента к субстрату.
2. Концентрация субстрата значительно больше Km ([S] >> Km). В этом случае величиной Km можно пренебречь, при решении получим, что скорость реакции максимальна (плато на графике).
3. Концентрация субстрата значительно меньше Km ([S]
Уравнение Михаэлиса — Ментен
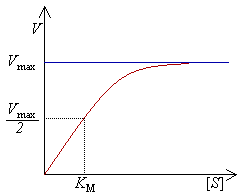
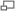
Диаграмvа скорости реакции V как функции от концентрации [S].
Уравне́ние Михаэ́лиса — Ме́нтен — основное уравнение ферментативной кинетики, описывает зависимость скорости реакции, катализируемой ферментом, от концентрации субстрата и фермента. Простейшая кинетическая схема, для которой справедливо уравнение Михаэлиса:
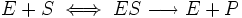
Уравнение имеет вид:
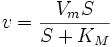 ,
,
· 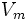 — максимальная скорость реакции, равная
— максимальная скорость реакции, равная 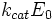 ;
;
· 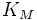 — константа Михаэлиса, равная концентрации субстрата, при которой скорость реакции составляет половину от максимальной;
— константа Михаэлиса, равная концентрации субстрата, при которой скорость реакции составляет половину от максимальной;
· 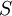 — концентрация субстрата.
— концентрация субстрата.
Вывод уравнения
Вывод уравнения был впервые предложен Бриггсом и Холдейном. Вывод уравнения скорости ферментативной реакции, описываемой схемой Михаэлиса-Ментен.
Обозначения констант скоростей:
k1 — константа скорости реакции образования фермент-субстратного комплекса из фермента и субстрата
k-1 — константа скорости реакции диссоциации фермент-субстратного комплекса на фермент и субстрат
k2 — константа скорости реакции превращения фермент-субстратного комплекса в фермент и продукт
Для фермент-субстратного комплекса применим метод квазистационарности, так как в подавляющем большинстве реакций константа скорости превращения фермент-субстратного комплекса в фермент и продукт много больше, чем константа скорости образования ферменто-субстратного комплекса из фермента и субстрата. Иными словами:
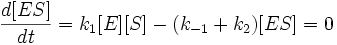
Учтем тот факт, что фермент, изначально находившийся только в свободной форме, в процессе реакции находится как в виде фермент-субстратного комплекса, так и в виде молекул свободного фермента. Таким образом:
Преобразуем это к виду:
И подставим в первое уравнение. После раскрытия скобок и группировки слагаемых получим следующее:
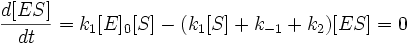
Выразим отсюда концентрацию фермент-субстратного комплекса:
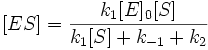
Скорость ферментативной реакции в целом (то есть скорость образования продукта) представляет собой скорость распада фермент-субстратного комплекса по реакции первого порядка с константой k2:
Подставим в эту формулу выражение, которое мы получили для концентрации ES. Получим:
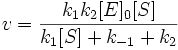
Разделим числитель и знаменатель на k1. В результате:
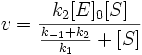
Выражение в знаменателе — (k-1+k2)/k1 — называется константой Михаэлиса (Km). Это кинетическая константа (с размерностью концентрации), которая равняется такой концентрации субстрата, при которой скорость ферментативной реакции составляет половину от максимального значения.
Для начальной стадии реакции можно пренебречь уменьшением концентрации субстрата. Тогда выражение для начальной скорости реакции будет выглядеть так:
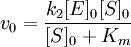
Если k-1>k2, то на первой стадии ферментативной реакции с течением времени устанавливается равновесие (квазиравновесный режим протекания реакции), и в выражение для скорости ферментативной реакции входит уже не константа Михаэлиса, а субстратная константа KS, характеризующая взаимодействие фермента с субстратом в равновесных условиях:
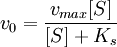 ;
; 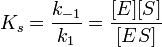
По значению KS можно судить о химическом сродстве субстрата к ферменту.
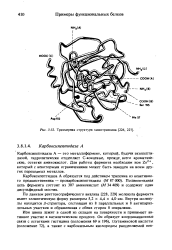
Металлоферменты и ферменты, активируемые металлами
Металлоферменты содержат определенное количество ионов металлов, имеющих функциональное значение и остающихся связанными с молекулой фермента в ходе его очистки. Ферменты, активируемые металлами, связывают последние менее прочно, но для своей активности требуют добавления металлов в среду. Таким образом, разграничение между металлоферментами и ферментами, активируемыми металлами, основано на сродстве данного фермента к иону «своего» металла. Механизмы, основанные на участии ионов металлов в катализе, в обоих случаях, по-видимому, сходны.
Тройные комплексы фермент—металл — субстрат
Для тройных (трехкомпонентных) комплексов, включающих каталитический центр (Enz), ион металла (М) и субстрат (S) со стехиометрией 1:1:1, возможны четыре различных схемы образования:
Enz—S—М М —Enz—S
Комплекс с мостиковым Комплекс с мостиковым субстратом ферментом
М
Enz—M—S Enz|
S
Простой комплекс с Циклический комплекс
мостиковым металлом с мостиковым металлом
В случае ферментов, активируемых металлами, реализуются все четыре схемы. Для металлофермен-тов образование комплекса Enz—S—М невозможно, иначе они не могли бы удерживать металл в процессе очистки (они находятся в форме Enz — — М). Можно сформулировать три общих правила.
1. Большинство (но не все) киназ (АТР:фосфо-трансферазы) образуют комплексы с мостиковым субстратом типа Enz—нуклеозид—М.
2. Фосфотрансферазы, использующие в качестве субстрата пируват или фосфоенолпируват, другие ферменты, катализирующие реакции с участием фосфоенолпирувата, а также карбоксилазы образуют комплексы с мостиковым металлом.
3. Данный фермент может быть способен к образованию мостикового комплекса одного типа с одним субстратом и другого типа — с другим.
Комплексы с мостиковым ферментом(М —Enz—S)
Металлы в комплексах с мостиковым ферментом, по-видимому, выполняют структурную роль, поддерживая активную конформацию (примером служит глутаминсинтаза), или образуют мостик с другим субстратом (как в пируваткиназе). В пиру-ваткиназе ион металла играет не только структурную роль, но и удерживает один из субстратов (АТР) и активирует его:
А
Пируваткиназа—АТР
Креатин.
Комплексы с мостиковым субстратом
Образование тройных комплексов с мостиковым субстратом, которое наблюдается при взаимодействии ферментов с нуклеозидтрифосфатами, по-видимому, связано с выстеснением Н20 из координационной сферы металла, место которой занимает АТР:
АТР- + M(H20) 2+ fi г± АТР—М(Н20) 2 \ + ЗН20.
Затем субстрат связывается с ферментом, образуя тройной комплекс:
АТР—М(Н20) 2 :, + Enz 2 :v
В фосфотрансферазных реакциях ионы металлов, как полагают, активируют атомы фосфора и образуют жесткий полифосфат-адениновый комплекс в соответствующей конформации, который включается в состав активного четырехкомпонентного комплекса.
Комплексы с мостиковым металлом
м
Enz-M-SnnnEnz |
S
Кристаллографические данные, а также анализ первичной структуры показывают, что в активных центрах многих белков в связывании металла участвует остаток гистидина (примерами служат кар-боксипептидаза А, цитохром с, рубредоксин, мет-миоглобин и метгемоглобин; см. гл. 6). Лимитирующей стадией образования бинарных (двухкомпо-нентных) комплексов Enz—М во многих случаях является вытеснение воды из координационной сферы иона металла. Активация многих пептидаз ионами металла является медленным процессом, длящимся несколько часов. Среди таких металлоферментов особое значение в живых системах имеют цитохромы и гемсодержащие ферменты, активными группировками которых служат железопорфириновые комплексы ( см. стр. [1]
Во многих металлоферментах, особенно катализирующих окислительно-восстановительные реакции, присутствуют именно металлы с переменной валентностью, действующие как акцепторы водорода, например железо, медь, марганец, молибден. Известно, что для активации таких ферментов, как ксантиноксидаза или нитроредуктаза, молибден должен быть в шестивалентном состоянии. [2]
Карбоксипептидаза — это металлофермент, содержащий один атом цинка на молекулу белка. Карбоксипептидаза катализирует гидролиз С-концевой пептидной связи в белках и олигопеп-тидах и сложных эфиров а-оксикислот. По данным рентгеноструктурного анализа Карбоксипептидаза представляет собой глобулярный белок, в котором содержится один атом цинка, координированный двумя остатками гистидина. Кроме того, в состав активного центра входят карбоксильная ( Glu-270), фенольная ( Туг-248) и гуанидиновая ( Arg-145) группы. [3]
Ингибирует широкий набор металлоферментов, включая гидроге-назу и гемоглобины ( при низких конц. СО-инги-бирование, обратимое под действием света, характерно для цитохромокси-дазы и др. гемсодержащих белков. Карбонилы Си и др. металлов не чувствительны к свету. Моноксид углерода сильно токсичен для многих живых тканей и микроорганизмов. [4]
СОД относится к металлоферментам, у которых в активном центре происходит восстановление и окисление иона металла. [5]
 | Трехмерная структура химотрипсина. |
Карбоксипептидаза А — это металлофермент, который, будучи экзопепти-дазой, гидролитически отщепляет С-концевые, прежде всего ароматические, остатки аминокислот. Для работы фермента необходим ион Zn2, который с некоторыми ограничениями может быть замещен на ионы других переходных металлов. [6]
Сравнительное изучение каталитических свойств различных металлоферментов в начале было главным образом ограничено оценкой их относительной каталитической активности. Однако детальное изучение катализа и связывания лиганда для серии металлоферментов может пролить свет на роль иона металла, если при отсутствии каталитической активности сохраняется связывание металла. [7]
Прочность связи фосфата с металлоферментом удивительно велика. Константа устойчивости комплекса, равная около 106 М 1 [51, 63, 76], выше, чем можно было бы ожидать для взаимодействия НРО f — и иона двухвалентного металла. Вероятно, вблизи иона цинка находится еще один катионный центр связывания, например протон аминогруппы боковой цепи аминокислоты, взаимодействующий с кислородными атомами фосфата. Образование фосфорилфермента при реакции с субстратом и НРО можно считать твердо установленным. После разрушения белка фосфат оказывается присоединенным к серину. Отсюда делается вывод о том, что в активном центре вблизи иона цинка находится остаток серина, ориентация которого делает возможной его атаку по фосфорильному атому присоединенного производного фосфата. Вполне вероятно, что фосфорильная группа, присоединенная к серину, сохраняет связь с ионом цинка. [8]
ЭКВ отчетливо проявляются в свойствах металлоферментов. Металлы служат кофакторами многих ферментов — в большинстве классов ферментов имеются металлозависимые. [9]
В части IV рассмотрены структуры металлоферментов и механизмы, посредством которых ионы металлов принимают участие в ферментативной активности, в частности, в разрыве связей. [10]
Малер [8] подчеркивает различия между металлоферментами, в которых металл прочно связан с белком ( по существу необратимо), и ферментами, активированными металлами, в которых связь металла с белком слаба и легкообратима. [11]
В настоящее время известно более 100 истинных металлоферментов, участвующих в большинстве реакций клеточного метаболизма. Многие из них передают электроны за счет металлов с переменной валентностью. В этом случае можно постулировать, что металл принимает непосредственное участие в осуществлении каталитического акта. [12]
Ферменты окислительно-восстановительного действия являются чаще всего металлоферментами, так как в качестве компонентов каталитически активных центров этих ферментов служат металлы. В истинных металлоферментах металл прочно связан в строго определенных стехиометрических соотношениях с теми или иными химическими группировками апофермента или со специальной группой небелковой природы. [13]
В этих же органах и тканях находятся соответствующие металлоферменты или ферменты, активируемые тем или иным металлом. Так, для меди, цинка, молибдена, селена, марганца критическим органом является печень; для марганца, кобальта — также и щитовидная железа; для кадмия и цинка — мужские половые железы и почки; для бериллия — костная ткань. Полагают также, что распределение металлов внутри клетки коррелирует с содержанием в ней металлосодержащих ферментов
Дата добавления: 2017-01-13 ; просмотров: 4683 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Кинетика ферментативных реакций
Одним из характерных проявлений жизни является удивительная способность живых организмов кинетически регулировать химические реакции, подавляя стремление к достижению термодинамического равновесия. Ферментативная кинетика занимается исследованием закономерностей влияния химической природы реагирующих веществ (ферментов, субстратов) и условий их взаимодействия (концентрация, рН среды, температуры, присутствие активаторов или ингибиторов) на скорость ферментативной реакции. Главной целью изучения кинетики ферментативных реакций является получение информации, которая может способствовать выяснению молекулярного механизма действия фермента.
Общие принципы кинетики химических реакций применимы и к ферментативным реакциям. Известно, что любая химическая реакция характеризуется константой термодинамического равновесия. Она выражает состояние химического равновесия, достигаемого системой, и обозначается Кр. Так, для реакции:
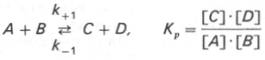
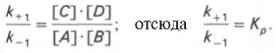
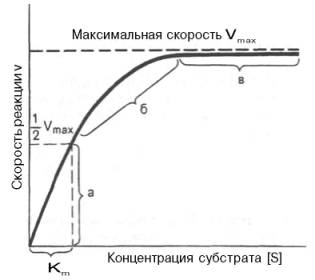
Рис. 4.12. Теоретический график зависимости скорости ферментативной реакции от концентрации субстрата при постоянной концентрации фермента.
а — реакция первого порядка (при [ S ]
Таким образом, константа равновесия равна отношению констант скоростей прямой и обратной реакций. Величину, обратную константе равновесия, принято называть субстратной константой, или, в случае ферментативной реакции, константой диссоциации фермент–субстратного комплекса, и обозначать символом KS. Так, в реакции
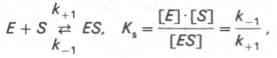
т.е. KSравна отношению произведения концентрации фермента и субстрата к концентрации фермент-субстратного комплекса или отношению констант скоростей обратной и прямой реакций. Следует отметить, что константа KSзависит от химической природы субстрата и фермента и определяет степень их сродства. Чем ниже значение KS, тем выше сродство фермента к субстрату.
При изучении кинетики ферментативных реакций следует учитывать одну важную особенность этих реакций (не свойственную обычным химическим реакциям), связанную с явлением насыщения фермента субстратом. При низкой концентрации субстрата зависимость скорости реакции от концентрации субстрата (рис. 4.12) является почти линейной и подчиняется кинетике первого порядка. Это означает, что скорость реакции S —> Р прямо пропорциональна концентрации субстрата S и в любой момент времени t определяется следующим кинетическим уравнением:
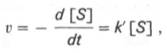
где [S] – молярная концентрация субстрата S; –d[S]/dt – скорость убыли субстрата; k’ – константа скорости реакции, которая в данном случае имеет размерность, обратную единице времени (мин –1 или с –1 ).
При высокой концентрации субстрата скорость реакции максимальна, становится постоянной и не зависящей от концентрации субстрата [ S ] . В этом случае реакция подчиняется кинетике нулевого порядка v = k» (при полном насыщении фермента субстратом) и целиком определяется концентрацией фермента. Различают, кроме того, реакции второго порядка, скорость которых пропорциональна произведению концентраций двух реагирующих веществ. В определенных условиях при нарушении пропорциональности говорят иногда о реакциях смешанного порядка (см. рис. 4.12).
Изучая явление насыщения, Л. Михаэлис и М. Ментен разработали общую теорию ферментативной кинетики. Они исходили из предположения, что ферментативный процесс протекает в виде следующей химической реакции:
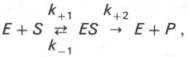
т.е. фермент Е вступает во взаимодействие с субстратом S с образованием промежуточного комплекса ES, который далее распадается на свободный фермент и продукт реакции Р. Математическая обработка на основе закона действующих масс дала возможность вывести уравнение, названное в честь авторов уравнением Михаэлиса–Ментен, выражающее количественное соотношение между концентрацией субстрата и скоростью ферментативной реакции:
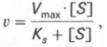
Из уравнения Михаэлиса–Ментен следует, что при высокой концентрации субстрата и низком значении KSскорость реакции является максимальной, т.е. v = Vmax(реакция нулевого порядка, см. рис. 4.12). При низкой концентрации субстрата, напротив, скорость реакции оказывается пропорциональной концентрации субстрата в каждый данный момент (реакция первого порядка).
Следует указать, что уравнение Михаэлиса–Ментен в его классическом виде не учитывает влияние на скорость ферментативного процесса продуктов реакции, например в реакции
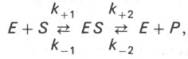
и носит несколько ограниченный характер. Поэтому были предприняты попытки усовершенствовать его. Так, было предложено уравнение Бриггса-Холдейна:
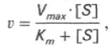
где Кm представляет собой константу Михаэлиса, являющуюся экспериментально определяемой величиной. Она может быть представлена следующим уравнением:
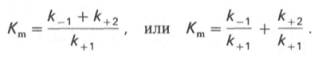
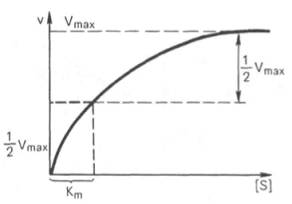
Рис. 4.13. Кривая уравнения Михаэли-са-Ментен: гиперболическая зависимость начальных скоростей катализируемой ферментом реакции от концентрации субстрата.
В числителе представлены константы скоростей распада комплекса ES в двух направлениях (в сторону исходных Е и S и в сторону конечных продуктов реакции Е и Р). Отношение k–1/ k+1представляет собой константу диссоциации ферментсубстратного комплекса KS, тогда:
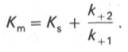
Отсюда вытекает важное следствие: константа Михаэлиса всегда больше константы диссоциации фермент-субстратного комплекса KSна величину
Для определения численного значения Кm обычно находят ту концентрацию субстрата, при которой скорость ферментативной реакции v составляет половину от максимальной Vmax, т.е. если v = 1 /2 Vmaх. Подставляя значение v в уравнение Бриггса–Холдейна, получаем:
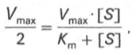
разделив обе части уравнения на Vmах, получим
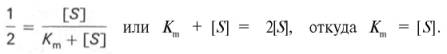
Таким образом, константа Михаэлиса численно равна концентрации субстрата (моль/л), при которой скорость данной ферментативной реакции составляет половину от максимальной.
Определение величины Кm имеет важное значение при выяснении механизма действия эффекторов на активность ферментов и т.д. Константу Михаэлиса можно вычислить по графику (рис. 4.13). Отрезок на абсциссе, соответствующий скорости, равной половине максимальной, будет представлять собой Кm.
Пользоваться графиком, построенным в прямых координатах зависимости начальной скорости реакции v0 от начальной концентрации субстрата [S0], неудобно, поскольку максимальная скорость Vmaxявляется в данном случае асимптотической величиной и определяется недостаточно точно.
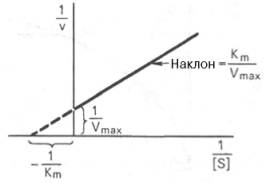
Рис. 4.14. График Лайнуивера-Бэрка.
Для более удобного графического представления экспериментальных данных Г. Лайнуивер и Д. Бэрк преобразовали уравнение Бриггса–Хол-дейна по методу двойных обратных величин исходя из того принципа, что если существует равенство между двумя какими-либо величинами, то и обратные величины также будут равны. В частности, если
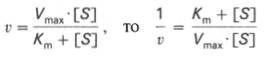
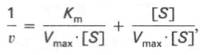
то после преобразования получаем уравнение:
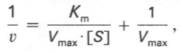
которое получило название уравнения Лайнуивера–Бэрка. Это уравнение прямой линии: у = ах + b. Если теперь в соответствии с этим уравнением построить график в координатах 1/v (y) от l/[S] (x), то получим прямую линию (рис. 4.14), тангенс угла наклона который будет равен величине Km/Vmax; отрезок, отсекаемый прямой от оси ординат, представляет собой l/Vmax(обратная величина максимальной скорости). Если продолжить прямую линию за ось ординат, тогда на абсциссе отсекается отрезок, соответствующий обратной величине константы Михаэлиса – 1/Кm (см. рис. 4.14). Таким образом, величину Кm можно вычислить из данных наклона прямой и длины отрезка, отсекаемого от оси ординат, или из длины отрезка, отсекаемого от оси абсцисс в области отрицательных значений.
Следует подчеркнуть, что значения Vmax, как и величину Кm, более точно, чем по графику, построенному в прямых координатах, можно определить по графику, построенному по методу двойных обратных величин. Поэтому данный метод нашел широкое применение в современной энзимологии. Предложены также аналогичные графические способы определения Кm и Vmaxв координатах зависимости v от v/[S] и [S]/v от [S].
Следует отметить некоторые ограничения применения уравнения Ми-хаэлиса–Ментен, обусловленные множественными формами ферментов и аллостерической природой фермента. В этом случае график зависимости начальной скорости реакции от концентрации субстрата (кинетическая
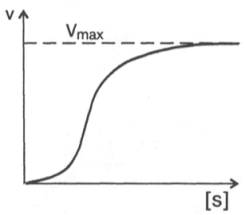
Рис. 4.15. Сигмоидная кинетическая кривая насыщения субстратом.
кривая) имеет не гиперболическую форму, а сигмоидный характер (рис. 4.15) наподобие кривой насыщения гемоглобина кислородом. Это означает, что связывание одной молекулы субстрата в одном каталитическом центре повышает связывание субстрата с другим центром, т.е. имеет место кооперативное взаимодействие, как и в случае присоединения кислорода к 4 субъединицам гемоглобина. Для оценки концентрации субстрата, при которой скорость реакции составляет половину максимальной, в условиях сигмоидного характера кинетической кривой обычно применяют преобразованное уравнение Хилла:
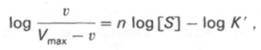
где К’ – константа ассоциации; n – число субстратсвязывающих центров.
http://helpiks.org/8-94053.html
http://xumuk.ru/biologhim/049.html