Ограниченный метод Хартри-Фока. Уравнения Рутана.
До сих пор развиваемая теория оставалась общей в том смысле, что не зависела от типа рассматриваемых молекул. Если считать, что в молекуле четное число электронов и число электронов со спином α равно числу электронов со спином β, т.е. Nα/2 = Nβ/2 = n. Кроме того, предположим, что все электроны спарены, т.е. пары электронов занимают одну и ту же пространственную орбиталь, различаясь только спином. Качество этого приближения ухудшается при растяжении связей и, наконец, нарушается при диссоциации связи, когда электронам энергетически выгоднее находиться в неспаренном состоянии. Такой тип молекул в действительности очень распространен и называется молекулами с замкнутыми (закрытыми) электронными оболочками. Тогда уравнения Хартри-Фока сводятся к более простому виду:
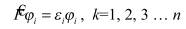
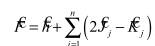
Уравнение Хартри-Фока, в принципе, позволяет найти волновые функции многоэлектронной системы, однако в случае молекул его прямое решение все еще оказывается весьма сложным. Для описания молекулярных систем требуется сделать дополнительные упрощения. Будем искать одноэлектронные функции многоэлектронной системы в виде линейной комбинации одноэлектронных функций центрированных на ядрах атомов (атомных орбиталей). Это приближение называется приближением «Молекулярная Орбиталь – Линейная Комбинация Атомных Орбиталей», или МО-ЛКАО.
Тогда можно показать, что выражение (2.46) можно представить в виде системы линейных уравнений (уравнения Рутана):
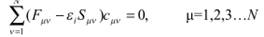
или в матричном виде:
FC=SCε,(2.51)
Здесь матрица F— т.н. оператор Фока(фокиан).Матрица Pназывается матрицей плотности, S –называются интегралами перекрывания, χi — базисные функции, центрированные на ядрах атомов, r— векторы пространственных координат электронов. Величины εi называются одноэлектронными энергиями или энергией молекулярной орбиталиi. Можно показать, что энергия МО с точностью до знака равна энергии удаления электрона с данной МО (т.е. потенциалу ионизации с данной МО) при условии, что орбитали системы в ходе этого процесса рассматриваются как неизменные (Теорема Кумпанса).
Решение уравнения Рутана (2.51) генерирует набор коэффициентов cij, которые являются коэффициентами разложения молекулярной орбитали φi (ri)по базисным функциям (атомным орбиталям) χ (ri).
Однако, уравнения Рутана имеют еще одну сложность — фокиан зависит от матрицы плотности, которая, в свою очередь определяется через cij — т.е. через решения самих уравнений Рутана. Чтобы выйти из этого заколдованного круга, В.А. Фок предложил решать уравнения (2.48) с помощью итерационной процедуры, которая получила название процедуры самосогласования. Сам метод Хартри-Фока часто называют методом самосогласованного поля (ССП). В соответствие с этой процедурой вначале выбирается некая приближенная матрица плотности и формируется начальный фокиан. Затем решаются уравнения (2.51) и находится новая (уже более точная) матрица плотности.
Процессу самосогласования посвящено большое количество специальных научных исследований. В настоящее время выработаны специальные алгоритмы (конвергеры), позволяющие ускорить сходимость процедуры ССП или добиться самосогласования для систем с плохой сходимостью. Наиболее простым методом является сдвиг виртуальных орбиталей на некоторую энергию dE вверх, так, что энергетическая щель между занятыми и виртуальными орбиталями увеличивается. Более сложным способом являются известные и часто применяемые процедуры DIIS и SOSCF, которые во многих случаях дают более высокое ускорение сходимости. Большинство современных квантовохимических программ позволяет произвольно использовать тот или иной конвергер по желанию пользователя. Следует, однако, помнить, что до настоящего времени нет универсальной процедуры, гарантирующей сходимость процедуры самосогласования во всех случаях.
Дата добавления: 2019-10-16 ; просмотров: 787 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
O Квантовая механика и квантовая химия (стр. 5 )
 | Из за большого объема этот материал размещен на нескольких страницах: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |


Уравнения Хартри-Фока также решаются методом самосогласованного поля, и полученные в результате орбитали называются каноническими. После вычисления канонических орбиталей могут быть получены и другие типы орбиталей – локализованные и гибридные.
Таким образом, метод Хартри-Фока представляет собой одноэлектронное приближение, для которого волновая функция представлена в виде одного определителя Слэйтера.
Недостатком метода Хартри-Фока является представление волновой функции в виде единственного детерминанта Слэйтера. Это не позволяет учесть взаимозависимого, или согласованного, движения электронов – электронной корреляции.
Модель Хартри-Фока является границей, разделяющей неэмпирические и полуэмпирические методы. Последние получаются при введении каких-либо дополнительных приближений. Решение Хартри-Фока может быть улучшено введением дополнительных детерминантов Слэйтера, что позволяет учесть электронную корреляцию. Таким образом, иерархию квантово-химических методов можно представить в следующем виде (рис. 3.3).


Р и с. 3.3. Иерархия квантово-химических методов
3.3. Выбор базисного набора,
уравнения Рутаана – Холла
Принципиально может быть выбран любой набор базисных функций, из которых формируется детерминант Слэйтера, – экспоненциальные, гауссовы функции, полиномы, объёмные и плоские волны и пр. При выборе базисных функций руководствуются следующими двумя правилами:
1) функции должны вести себя так, чтобы соответствовать физической модели, чтобы при увеличении базисного набора итерационную процедуру самосогласования можно было производить относительно быстро; т. е. функции должны стремиться к нулю при увеличении расстояния между ядрами и электронами до бесконечности;
2) выбранные функции должны позволять легко вычислять все необходимые интегралы.
Первый критерий предполагает предпочтительное использование экспоненциальных функций, локализованных на ядрах, например, функций водородоподобного атома или функций Слэйтера. Но, к сожалению, расчёт интегралов этих функций достаточно трудоёмок. Гауссовы функции достаточно просто позволяют вычислять интегралы, но они достаточно плохо описывают электронную структуру. Однако простота вычисления интегралов в данном случае имеет большее значение.
Гауссовы функции отличаются от функций Слэйтера в своей радиальной части (ξ и α – экспоненциальные множители):
– радиальная часть функции Слэйтера 
– радиальная часть функции Гаусса  .
.
Каждую молекулярную орбиталь можно представить в виде разложения по базисным орбиталям. Если в качестве базисных функций взять атомные орбитали (M функций в (3.3.1)), то молекулярные орбитали будут являться линейными комбинациями атомных орбиталей (МО = ЛКАО):

Уравнения Хартри – Фока при этом приобретают следующий вид:

Умножение слева на базисную функцию и интегрирование даёт уравнения Рутаана – Холла (3.3.3) (для систем с закрытой оболочкой). Фактически это уравнения Фока в базисе атомных орбиталей:
где 
Матрица S содержит элементы перекрывания между атомными орбиталями, матрица F – элементы матрицы Фока. Каждый матричный элемент Fij (3.3.4) содержит две чисти оператора Фока: 1) интегралы, соответствующие одноэлектронному оператору; 2) сумму по занятым МО произведений коэффициентов на двухэлектронные интегралы, соответствующие оператору электрон-электронного отталкивания. Последние можно представить как произведение матрицы плотности  (3.3.5) и матрицы двухэлектронных интегралов:
(3.3.5) и матрицы двухэлектронных интегралов:




где  – элементы матрицы плотности. (3.3.5)
– элементы матрицы плотности. (3.3.5)
Выражение (3.3.6) – более компактная форма матричного элемента матрицы Фока:

Общая энергия в терминах интегралов базисных функций принимает следующий вид (3.3.8):
 или (3.3.9), (3.3.10):
или (3.3.9), (3.3.10):


При решении уравнений Рутаана – Холла определяют собственные значения и собственные вектора матрицы Фока. Но для построения матрицы Фока должны быть известны коэффициенты разложения МО по АО. А поскольку они первоначально не известны, то нужно выбрать какие-то начальные коэффициенты для построения матрицы Фока. После выбора начальных коэффициентов строится матрица Фока и вычисляются её собственные значения (орбитальные энергии) и собственные векторы (молекулярные орбитали). Собственные векторы далее используются для построения матрицы Фока, и процедура повторяется. Вычисления повторяют до тех пор, пока значения собственных векторов не перестанут изменяться в пределах определённой точности. Полученный набор коэффициентов (собственных векторов) является решением в методе самосогласованного поля Хартри – Фока.
Матрица Фока и, следовательно, полная энергия системы зависит только от занятых МО. При решении уравнений Рутаана – Холла мы используем М базисных функций, из которых N функций занято электронами. Остальные (M – N) функций не заняты электронами и называются виртуальными МО. Виртуальные МО ортогональные ко всем занятым МО и не имеют прямой физической интерпретации. Энергии занятых орбиталей, в соответствии с теоремой Купманса, равны потенциалам ионизации.
Для построения матрицы Фока необходимо вычислить ряд интегралов:
1) интегралы от двух функций и одноэлектронного оператора  (для М базисных функций потребуется вычислить
(для М базисных функций потребуется вычислить  таких интегралов) – эти одноэлектронные интегралы называют óстовными, и они описывают взаимодействие электрона со всем набором ядер;
таких интегралов) – эти одноэлектронные интегралы называют óстовными, и они описывают взаимодействие электрона со всем набором ядер;
2) интегралы от четырёх функций и двухэлектронного оператора  ; их количество пропорционально
; их количество пропорционально  , они называются двухэлектронными. Для двухэлектронных интегралов 4 базисные функции могут быть локализованы на 1, 2, 3 или 4 различных атомных центрах. Соответствующие интегралы называются одно-, двух-, трех — и четырёхцентровыми. Подобные интегралы гораздо проще вычисляются при использовании функций Гаусса.
, они называются двухэлектронными. Для двухэлектронных интегралов 4 базисные функции могут быть локализованы на 1, 2, 3 или 4 различных атомных центрах. Соответствующие интегралы называются одно-, двух-, трех — и четырёхцентровыми. Подобные интегралы гораздо проще вычисляются при использовании функций Гаусса.
Таким образом, формально сложность нахождения решений уравнений Рутаана – Холла пропорциональна четвёртой степени размера базиса ().
С увеличением числа базисных функций точность вычисления МО улучшается. В предельном случае, при использовании бесконечного базиса, получается результат, идентичный численному решению уравнений Хартри – Фока. Этот результат называется пределом теории Хартри – Фока. Но этот предел не является точным решением уравнения Шрёдингера. Хартри – фоковский предел соответствует лишь наилучшей однодетерминантной волновой функции.
3.4. Ограниченный и неограниченный методы
Хартри – Фока
Слэйтеровский детерминант может быть построен из спин-орбиталей, являющихся произведением пространственной функции на спиновую (α или β). Если не накладывается никаких ограничений на формы спин-орбиталей, то пробная волновая функция называется неограниченной, а соответствующий метод – неограниченным методом Хартри – Фока (Unrestricted Hartree—Fock, UHF).
Если мы исследуем систему с чётным числом электронов и синглетной волновой функцией, что справедливо для систем с замкнутой оболочкой, то на волновую функцию накладывается определённое ограничение. Оно заключается в том, что на каждой орбитали должно располагаться два электрона с разными спинами. Подобная волновая функция называется ограниченной, а соответствующий метод – ограниченным методом Хартри – Фока (Restricted Hartree-Fock, RHF).
Молекулы с открытыми оболочками (имеющие нечётное количество электронов) также можно описывать с помощью ограниченных функций. Для дважды занятых орбиталей используется по одной функции для каждой пары электронов с противоположными спинами. Неспаренный электрон располагается на одной из орбиталей. Такой метод называется ограниченным методом Хартри – Фока для открытых оболочек (Rrestricted Open-shell Hartree—Fock, ROHF). Для частиц с открытыми оболочками метод UHF даёт хорошо определённые орбитальные энергии, которые можно рассматривать как ионизационные потенциалы. В методе ROHF невозможно преобразовать матрицу множителей Лагранжа ( ) в диагональную. Это значит, что орбитальные энергии, полученные методом ROHF, не являются хорошо определёнными, и их нельзя приравнивать к потенциалу ионизации, пользуясь теоремой Купманса.
) в диагональную. Это значит, что орбитальные энергии, полученные методом ROHF, не являются хорошо определёнными, и их нельзя приравнивать к потенциалу ионизации, пользуясь теоремой Купманса.
Уравнения хартри фока решаются численно
Наиболее удобным, с точки зрения квантовой химии, является такой метод расчета структуры и свойств молекул, который использовал бы информацию только о конфигурации электронных оболочек атомов, составляющих систему. Реализация такого метода позволила бы исследователям предсказывать существование и свойства новых материалов, еще не полученных в эксперименте, например, сверхтвердых материалов и др. [1].
Ab initio , т. е. первопринципные методы, используют вышеописанный принцип, однако из-за высокой сложности расчета в них также применяются некоторые приближения, которые не позволяют применить эти методы к любым системам. При этом точность расчета в большинстве случаев довольно высока. Так, например, в [2] были успешно предсказаны структура и свойства внутреннего ядра Земли, состоящего в основном из железа.
К основным методам расчета из первых принципов можно отнести следующие:
- метод Хартри -Фока и его дальнейшие развития;
- метод функционала электронной плотности .
1.1.1. Метод Хартри
1.1.1.1. Уравнение Хартри
Метод Хартри Фока, или метод самосогласованного поля, является одним из эффективных методов решения задач квантовой химии. Его идея состоит в том, что взаимодействие электрона с его окружением заменяется взаимодействием с неким усредненным полем . Таким образом, не решаемая квантовомеханическая задача многих тел сводится к решению одночастичного уравнения [1] (где Δ оператор Лапласа).
Для начала запишем уравнение Шредингера:
где Ψ многоэлектронная волновая функция рассматриваемой системы.
В теории Хартри многоэлектронная волновая функции представлялась в виде простого произведения одноэлектронных волновых функций
где ψ p ( ξ i ) нормированная одноэлектронная волновая функция для i -го электрона, находящегося на p -й орбитали, ξ i обобщенные координаты, включающие в себя пространственную и спиновую часть. Уравнение (1.2), конечно же, не отражает действительную ситуацию, поскольку электроны являются фермионами, а волновая функция (1.2) не подчиняется принципу Паули. Слэтер ( Slater ) в [3] видоизменил вид (1.2) и переписал волновую функцию в виде следующего детерминанта:
Записанная в таком виде волновая функция подчиняется принципу Паули. Видно, что если два электрона обладают одинаковыми обобщенными координатами, общая волновая функция системы становится равной нулю. Одноэлектронные волновые функции в (1.3) также включают в себя спиновую часть.
Если система состоит только из спаренных электронов (система с замкнутыми оболочками closed — shell system ), то уравнение (1.3) удобно переписать в следующем виде:
где α и β обозначают спины, направленные вверх и вниз соответственно [4].
Системы с нечетным числом электронов являются системами с незамкнутыми (открытыми) оболочками ( open — shell system ). В этом случае в уравнении волновой функции (1.4) каждой спиновой координате соответствует своя пространственная координата.
В случае нормированной волновой функции Ψ энергию системы можно выразить следующим образом:
Для построения уравнений Хартри Фока рассмотрим двухэлектронный атом с зарядом ядра Z , в этом случае многоэлектронная функция (1.3) будет иметь следующий вид:
Выражение (1.5), таким образом, может быть записано в следующем виде:
Гамильтониан двухэлектронного атома включает в себя следующие члены:
где Hi гамильтониан взаимодействия i -го электрона с ядром, оператор электростатического взаимодействия между первым и вторым электронами.
Энергия двухэлектронного атома в соответствии с (1.5) будет равна
Рассмотрим каждый член выражения (1.9) в отдельности. Для члена можно записать
Преобразуем это выражение и выделим в нем интегралы по координатам индивидуальных электронов. Учитывая при этом ортонормированность одноэлектронных волновых функций, получим
Подобное выражение получается для второго члена из (1.9):
Учитывая тождественность электронов, видим, что первый и второй члены в (1.11) равны соответственно первому и второму членам в (1.12). Следовательно,
Аналогично можно найти, что
В случае последнего слагаемого в (1.9) нельзя разделить на одноэлектронные члены. Вычисляя его ожидаемое значение с антисимметричной волновой функцией и делая очевидные преобразования, получим
Подставляя выражения (1.13) (1.15) в (1.9), находим для ожидаемого значения гамильтониана
Первый и второй члены в этом выражении описывают кинетическую энергию электронов, находящихся на орбиталях ψ 1 и ψ 2 соответственно, третий и четвертый
взаимодействия этих электронов с ядром, пятый член (кулоновский интеграл) описывает электростатическое отталкивание между двумя электронами, один из которых находится на орбитали ψ 1, а второй на орбитали ψ 2. Шестой член описывает обменное взаимодействие (обменный интеграл). Распределение заряда для первого электрона описывается произведением , аналогичное выражение дает распределение заряда для второго электрона. Этот член обусловливает различие в энергии между синглетным и триплетным состояниями двухэлектронной системы.
Функция ψ μ ( ξ i ), как уже было сказано выше, включает в себя пространственные и спиновые координаты i -го электрона. Оператор воздействует только на пространственные координаты. Поэтому вследствие ортогональности различных спиновых функций обменный интеграл отличается от нуля только в том случае, если две входящие в него спиновые функции совпадают. Этот интеграл всегда положителен, поскольку описывает электростатическое отталкивание, но он входит в выражение (1.16) с отрицательным знаком. Следовательно, если спины электронов одинаковы, то вычисленная энергия двухэлектронного атома при заданных пространственных частях функций ψ 1 и ψ 2 оказывается ниже, чем в случае, когда спины электронов различаются. Триплетные состояния соответствуют наличию у электронов одинакового спина. И поэтому из двух спиновых состояний, возникающих в двухэлектронной системе, они характеризуются более низкой энергией, что согласуется с правилом Хунда.
Обобщим (1.16) на случай многоэлектронного атома. В обобщенное выражение должны войти члены, которые описывают кинетическую энергию и притяжение электронов к ядрам и включают одноэлектронную волновую функцию на каждой занятой спин-орбитали и, кроме того, кулоновский и обменный интегралы для каждой пары электронов. Результирующее выражение имеет вид
Выражение (1.17) служит отправной точкой для получения уравнений самосогласованного поля Хартри Фока. Процедура их вывода заключается в том, чтобы минимизировать выражение (1.17) путем варьирования орбиталей, соблюдая при этом требование, чтобы одноэлектронные орбитали были ортонормированы. Используем для этого метод множителей Лагранжа, позволяющий находить экстремум функции многих переменных (см. п риложение 1). Варьируемая величина представляется в виде суммы рассматриваемой функции и произведений каждого ограничительного условия на неопределенный (постоянный) множитель. Вариация этой суммы считается равной нулю. В данном случае ограничительными условиями являются требования нормированности каждой орбитали и ортогональности каждой пары орбиталей. Таким образом, варьируемую величину следует записать в виде , где λ μ ν множители Лагранжа. Варьируя это выражение по одной из орбиталей, например по ψ μ ( ξ 1), получим
В уравнении (1.19) явно выписанные члены получаются при наличии функции ψ μ в левой части интеграла, а соответствующие комплексно-сопряженные члены появляются, если функция ψ μ оказывается в правой части интеграла. В результате суммирования по μ остается только один член, содержащий индекс μ . Суммирование по ν остается и не распространяется только на члены . Волновые функции можно сконструировать так, что из этих членов останутся только те, в которых ν = μ .
Теперь, чтобы минимизировать рассматриваемую функцию, следует принять, что выражение (1.19) равно нулю. Если сумма функции и комплексно-сопряженной ей величины должна быть равна нулю, то каждая из них порознь тоже должна быть равна нулю. Следовательно, можно записать
Запишем третий член уравнения в интегральной форме:
Умножив и разделив выражение (1.21) на произведение , получим:
где обменный оператор. Если выражение назвать кулоновским оператором, обозначив его символом , то уравнение (1.20) можно переписать в виде:
Заменив величину λ μ μ на ε μ , а сумму операторов в скобках на (оператор Фока или фокиан), получим псевдоуравнение (нелинейное уравнение) на собственные значения:
Оператор Фока можно построить для каждой занятой одноэлектронной орбитали системы. Псевдоуравнения на собственные значения могут быть решены для каждой орбитали. Однако оператор Фока содержит операторы и , зависящие от распределения всех электронов, кроме того электрона, который описывается данным уравнением на собственные значения. Такое уравнение необходимо решать с помощью итерационной процедуры самосогласования [5].
В случае системы с замкнутыми электронными оболочками оператор Фока имеет следующий вид:
1.1.1.2. Базисные функции
На практике для решения уравнения (1.24) необходимо представлять молекулярную орбиталь (волновую функцию исследуемой системы) в виде комбинации атомных орбиталей (АО) (волновых функций атомов, входящих в систему), которые в свою очередь представимы как линейная комбинация конечного числа базисных состояний χ p :
Выбор базисных атомных функций является важной задачей, так как именно он определяет, насколько точно разложение (1.26) аппроксимирует молекулярную орбиталь Хартри Фока. Этот ряд должен достаточно быстро сходиться, т. е. малое число атомных орбиталей должно аппроксимировать молекулярную орбиталь с требуемой точностью. Существует три основных критерия для выбора базисных функций:
1. Базисные функции должны давать в основном хорошее приближение к истинной волновой функции (например, возле ядер и на больших расстояниях от них).
2. Базисные функции должны допускать аналитическое вычисление нужных интегралов.
3. Полное число базисных функций не должно быть очень большим [6].
На данный момент широко используются следующие наборы базисных функций:
Ø плоские волны ;
Ø слэтеровские орбитали ;
Ø гауссовы орбитали ;
Ø численные орбитали, форма которых оптимизируется из атомных расчетов.
Разложение по плоским волнам описывает обычно с хорошей точностью расчет электронной структуры периодичной кристаллической структуры. Также это разложение может быть успешно применено для расчета аморфных тел и/или конечных систем, таких как атомные кластеры или структуры с дефектами. С математической точки зрения, только плоские волны формируют полный базисных набор, т.е. при увеличении числа базисных функций точность решения уравнения Фока также будет увеличиваться.
Херрингом ( Herring ) [7] предложен метод ортогонализованных плоских волн (ОПВ) Orthogonalized Plane Waves ( OPW ). Он обратил внимание на тот факт, что медленная сходимость ряда (1.26) связана с сильными осцилляциями волновых функций электронов проводимости в области сердцевины иона (см. также метод псевдопотенциала). В этой области волновые функции очень похожи на атомные волновые функции валентного электрона. Чтобы воспроизвести такие осцилляции в методе плоских волн, необходимо было бы строить разложение из огромного числа ( ) плоских волн. Сходимость можно улучшить, только если каким-то образом учесть эти осцилляции в самом базисе, по которому мы будем разлагать функции.
Именно это и сделал Херринг, воспользовавшись тем, что волновые функции, которые требуется найти, должны быть ортогональны волновым функциям внутренних оболочек (последние считаются известными). Таким образом, полное разложение для волновых функций зоны проводимости можно получить, если пользоваться не просто плоскими волнами, а плоскими волнами, которые предварительно были сделаны ортогональными к волновым функциям внутренних оболочек. В процессе ортогонализации учитываются осцилляции в области сердцевины ионов, что позволяет в дальнейшем достаточно хорошо описать и соответствующие осцилляции в волновых функциях, которые мы ищем. Следовательно, этот метод очень похож на метод плоских волн, но только в нем вместо обычных плоских волн фигурируют ортогонализованные плоские волны.
Запишем ортогонализованную плоскую волну в виде
где нормированная собственная функция внутренних оболочек, индекс t характеризует состояние внутренних оболочек (1 s , 2 s , 2 p , …), а индекс j указывает на положение иона; Ω нормированный объем (объем кристалла).
В том, что ОПВ действительно ортогональны функциям внутренних оболочек, можно убедиться, если умножить выражение для них слева на какую-нибудь из этих функций и проинтегрировать. Интеграл оказывается равным нулю, если мы предположим, что волновые функции внутренних оболочек, относящиеся к различным ионам, не перекрываются (что на самом деле является хорошей аппроксимацией), а также учтем ортогональность таких функций, отвечающих различным состояниям одного иона. Таким образом, они образуют полную систему для разложения функций зоны проводимости, и именно это важно.

Рис. 1-1. Схематическое изображение ячеечного потенциала [9]. Каждая яма вблизи центра уходит в ∞ , но на рисунке они обрезаны.
Несколько ранее Слэтер [8] предложил в качестве базиса для разложения волновых функций другой тип функций так называемые присоединенные плоские волны (ППВ) ( augmented plane waves APW ). Прежде чем начать конструировать эти функции, целесообразно сначала выбрать какую-то аппроксимацию для потенциала, который будет использоваться в расчетах. Можно ожидать, что вблизи каждого ядра потенциал будет, скорее всего, сферически симметричным (радиус сфер должен быть достаточно малым, чтобы потенциалы, отвечающие различным атомам, не перекрывались), а в пространстве между сферами положим потенциал равным некоторой константе (рис. 1-1).
Присоединенные плоские волны определяются следующим образом: в пространстве между сферами волновые функции представляют собой плоские волны. В интересующей нас области энергий мы строим также решения для сферически симметричного потенциала. Коэффициенты перед этими функциями подбираются таким образом, чтобы волновая функция на поверхности сферы, переходя в плоскую волну, не испытывала скачка. Однако избежать таким образом разрывности первой производной не удается. Собственные функции электронов, отвечающих данной энергии, могут быть точно разложены по таким ППВ. При этом ППВ заменяют в расчетах плоские волны или ОПВ. Как и в случае ОПВ, требуется лишь ограниченное число членов суммы, и поэтому для расчета энергетических зон метод ППВ является очень эффективным [9].
Рассмотрим теперь базисы гауссовых и слэтеровских орбиталей, которые применяется, в основном, для расчета конечных тел кластеров и отдельных молекул.
Слэтеровские орбитали являются наиболее естественными, если расчет начинается с изолированных атомов, так как функции слэтеровского типа являются точным решением водородоподобного атома. Однако некоторые интегралы кулоновского типа не могут быть решены аналитически при использовании этого набора.
В квантово-химических расчетах большее распространение получили простейшие виды атомных орбиталей слэтеровского типа АО Слэтера Зенера
нормированные вещественные сферические гармоники, где
присоединенные полиномы Лежандра, δ m равно 2 при m = 0 и равно 1 в остальных случаях.
Параметрами орбиталей (1.28) являются квантовые числа n, l и m, показатели экспоненты ζ и координаты точек центрирования орбиталей. Обычно для базисных функций используются квантовые числа, соответствующие занятым и ближайшим возбужденным состояниям электрона в атоме. В целях сокращения количества молекулярных интегралов можно включать в базисный набор только орбитали с главными квантовыми числами, не превосходящими главного квантового числа n для заполненных оболочек в атоме (минимальный базис). В ряде случаев (полуэмпирические методы) в базис включаются только валентные орбитали (валентное приближение).
Для определения ζ Слэтер в 1930 году предложил набор эмпирических правил, основанных на стремлении к наилучшему воспроизведению первых потенциалов ионизации атомов. Согласно этим правилам
где Z заряд ядра, n* эффективное (возможно, дробное) главное квантовое число, S константа экранирования, определяемая следующим образом.
1. Все электроны в атоме делятся на группы: 1s; 2s, 2p; 3s; 3p; 3d; 4s, 4p; 4d; 4f; и т. д.
2. S равно сумме следующих вкладов:
· 0 для любых электронов, расположенных во внешних группах;
· 0,35 от каждого электрона в группе, кроме рассматриваемого (0,30 в случае 1s-группы);
· для s- и p-электронов по 0,85 от каждого электрона с главным квантовым числом, на единицу меньшим, чем у рассматриваемого, и по 1,00 от каждого электрона следующих внутренних оболочек;
· для d — и f -электронов по 1,00 от каждого электрона, расположенного во внутренних группах.
Радиальные функции слэтеровского типа недостаточно точно описывают поведение атомных орбиталей Хартри Фока на небольших расстояниях от ядра. Этот недостаток устраняется аппроксимацией каждой из этих АО, по крайней мере, двумя слэтеровскими функциями с разными орбитальными экспонентами
Функции (1.32) называются дубль-зета-функциями, а соответствующий базисный набор дубль-зета-базисом (DZ, широкое применение имеет также трипл-зета-базис
TZ).
Квантово-химические расчеты с использованием слэтеровских функций в качестве базисных орбиталей являются достаточно сложными и трудоемкими. Гауссовы орбитали частично решают эту проблему, но иногда при этом разложении также необходимо достаточно большое число членов в ряду (1.26).
В 1950 году С. Ф. Бойс предложил использовать базисные функции гауссова типа, имеющие вид
Их основное отличие от слэтеровских функций (1.28) заключается в квадратичной зависимости от r аргумента экспоненты. Это позволяет представлять произведения гауссовых функций (центрированных в разных точках), встречающиеся в многоцентровых интегралах, в виде линейной комбинации одиночных гауссовых функций, центрированных в общем случае в другой точке пространства. В результате сильно упрощаются вычисления всех интегралов.
Одна слэтеровская АО аппроксимируется обычно несколькими гауссовыми функциями. Поэтому базис гауссовых функций всегда больше базиса слэтеровских АО (Рис. 1-2).
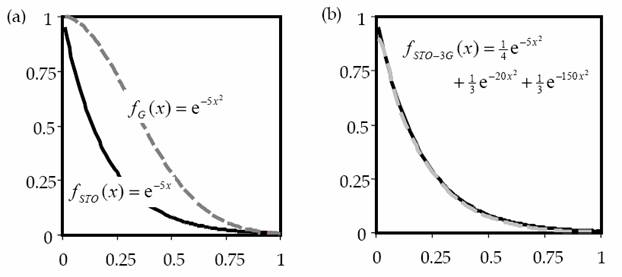
Рис. 1-2. Сравнение экспоненциальной и гауссовой функции (а) и той же экспоненциальной функции и комбинации трех гауссовых функций [10] ( b ).
Наиболее простым типом базисных наборов является ОСТ-nГФ (STO-nG), где каждая атомная орбиталь состоит из суммы n (обычно от двух до шести) функций гауссова типа, причем коэффициенты гауссовых функций подобраны таким образом, чтобы их линейные комбинации приближенно описывали поведение орбиталей слэтеровского типа. Проведение тестовых расчетов с использованием этих базисных наборов показало, что при n ≥ 3 результаты расчетов очень схожи. Поэтому наиболее широкое распространение получил минимальный базисный набор ОСТ-3ГФ (STO-3G). Минимальные базисные наборы включают только атомные орбитали, которые необходимы для размещения электронов нейтрального атома. Сферическая симметрия атомов и пространственная инвариантность молекул требуют включение всех трех типов np-орбиталей при появлении хотя бы одного p-электрона.
Малый размер и простота базиса ОСТ-3ГФ являются основными причинами его недостатков (неудовлетворительные результаты расчетов соединений электроположительных элементов третьего периода, переоценка стабильности малых циклов и π-акцепторной способности электроположительных элементов второго периода), которые устраняются при использовании более широких валентно-расщепленных и биэкспоненциальных базисов (DZ). В этих базисах АО составлены из двух частей внутренней, более компактной, и внешней более диффузной. При построении МО в процессе ССП коэффициенты каждой из орбиталей этих двух типов можно варьировать независимо.
В валентно-расщепленных базисных наборах на компактную и диффузную составляющие разделены только валентные орбитали. Поэтому схема записывается как m-npГФ (m-npG), где m число гауссовых функций, заменяющих каждую внутреннюю АО, n и p число гауссовых функций с разными значениями экспонент, аппроксимирующих каждую валентную АО. Наиболее часто применяемым является базис 6-31ГФ (6-31G). В биэкспоненциальных базисах (как указывалось выше) расщеплены как валентные, так и внутренние орбитали остова.
Для улучшения описания молекулярных орбиталей на больших расстояниях от ядра часто используют базисные наборы с включением поляризационных функций. Поляризационные функции дополнительные волновые функции с l + 1, где l орбитальное квантовое число последних заполненных атомных орбиталей. Одними из наиболее используемых поляризационных базисных наборов являются 6-31ГФ* и 6-31ГФ**, где звездочка обозначает добавление поляризационных d-функций к p-элементам (или f -функций к d -элементам). Вторая звездочка обозначает добавление поляризационных p-функций к 1s-орбиталям атомов водорода.
Для расчетов молекул, требующих более точного описания несвязывающих электронных пар, в базисные наборы вводятся специальные диффузные s- и p-функции со значениями экспонент от 0,1 до 0,01. Их включение обозначается знаком «+», например
3-21+ГФ.
Выбор базиса, подходящего для решения задачи, определяется стремлением получить более точное решение, с одной стороны, и ограничениями, связанными с ресурсами ЭВМ, с другой. Поэтому на практике применяется часто схема, когда полная оптимизация выполняется с использованием небольших базисов, после чего в более широких базисах проводятся расчеты для одной геометрической конфигурации и устанавливаются поправки, связанные с учетом электронной корреляции [11].
1.1.1.3. Уравнения Хартри
Базисные функции гауссова и слэтеровского типа, локализованные на разных атомах, не ортогональны друг другу:
з десь R 1 и R 2 радиус-векторы первого и второго атомов.
Пусть в качестве базисного набора выбран ряд таких неортогональных функций. Уравнение Фока будет записано в следующем виде:
Домножив это уравнение на и проинтегрировав, получим (в матричной форме записи)
где F матрица оператора Фока, S матрица перекрытия для базисных функций, определяемые следующим образом:
Часто уравнение (1.36) записывают в следующем виде:
Обобщенное уравнение на собственные значения (1.36) или (1.38) называется уравнением Рутана ( Ruthan ).
Матрица оператора Фока в случае системы с замкнутыми оболочками, таким образом, имеет следующий вид:
Индекс k обозначает орбиталь k , а p , q , r , s базисные функции. Суммирование по k идет по всем занятым орбиталям.
Введем следующее обозначение:
Prs называется матрицей плотности. Таким образом, матрица Фока может быть переписана в следующем виде:
С помощью унитарного преобразования обобщенное уравнение на собственные значения может быть приведено к обычному уравнению на собственные значения
и матрица V трансформирует S в единичную матрицу:
1.1.1.4. Алгоритм расчета с помощью метода Хартри
На следующей блок-схеме представлен алгоритм расчета с помощью метода Хартри Фока (рис. 1-3).
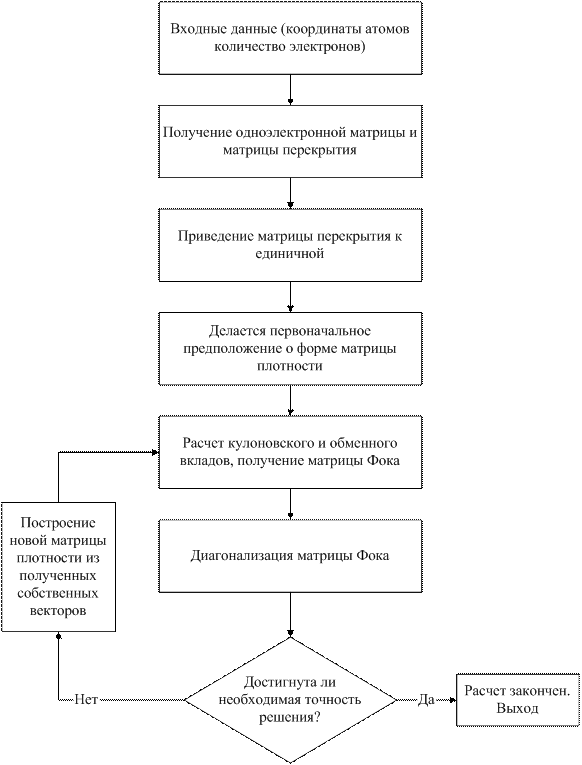
Рис. 1-3. Блок-схема алгоритма расчета с помощью метода Хартри Фока.
Входные данные: В качестве входящих данных задаются координаты атомов, заряды ядер, полное число электронов и базисные функции.
Получение матриц: Получение матриц, не зависящих от собственных векторов (матрица перекрытия Spq , матрица одноэлектронного гамильтониана Hpq , двухэлектронные интегралы ).
Приведение матрицы перекрытия к единичной: Расчет матрицы Vpq (см. (1.45)).
Диагонализация матрицы Фока: Используя матрицу Vpq , решается обобщенное уравнение на собственные значения (1.36).
Построение новой матрицы плотности: Используя полученные собственные векторы, строится новая матрица плотности [12].
http://pandia.ru/text/80/109/4005-5.php
http://test.kirensky.ru/master/articles/monogr/Book/Chapter_1_2.htm