Название: Реальные газы Раздел: Биология и химия Тип: курсовая работа Добавлен 05:48:01 05 апреля 2008 Похожие работы Просмотров: 5002 Комментариев: 20 Оценило: 5 человек Средний балл: 4.6 Оценка: неизвестно Скачать
Курсовая работа выполнил: студент 462 группы Махорт Александр
ГОУ ВПО Тюменский государственный университет
Понятие реального газа. Его свойства.
Газы (французское gaz; название предложено голланским учёным Я. Б. Гельмонтом), агрегатное состояние вещества, в котором его частицы не связаны или весьма слабо связаны силами взаимодействия и движутся свободно, заполняя весь предоставленный им объём. Вещество в газообразном состоянии широко распространено в природе. Газы образуют атмосферу Земли, в значительных количествах содержатся в твёрдых земных породах, растворены в воде океанов, морей и рек. Солнце, звёзды, облака межзвёздного вещества состоят из газов — нейтральных или ионизованных (плазмы). Встречающиеся в природных условиях газы представляют собой, как правило, смеси химически индивидуальных газов.
Газы обладают рядом характерных свойств. Они полностью заполняют сосуд, в котором находятся, и принимают его форму. В отличие от твёрдых тел и жидкостей, объём газа существенно зависит от давления и температуры. Коэффициент объёмного расширения газа в обычных условиях (0-100°С) на два порядка выше, чем у жидкостей, и составляет в среднем 0,003663 град-1.
Модель идеального газа, используемая в молекулярно-кинетической теории газов, позволяющая описывать поведение разрежённых реальных газов при достаточно высоких температурах и низких давлениях. При выводе уравнения состояния идеального газа размерами молекул и их взаимодействием друг с другом пренебрегают. Повышение давления приводит к уменьшению среднего расстояния между молекулами, поэтому необходимо учитывать объём молекул и взаимодействие между ними. При высоких давлениях и низких температурах указанная модель идеального газа непригодна.
Неидеальность газов в молекулярно-кинетической теории рассматривается как результат взаимодействия молекул. В первом приближении ограничиваются рассмотрением парных взаимодействий, во втором-тройных и т.д. Такой подход приводит к вириалъному уравнению состояния, коэффициенты которого могут быть теоретически рассчитаны, если известен потенциал межмолекулярных взаимодействий. Наиболее полезно вириальное уравнение при рассмотрении свойств газов малой и умеренной плотности. Этот вопрос будет раскрыт немного позже.
Наличие межмолекулярных взаимодействий оказывает влияние на все свойства реальных газов, в т.ч. приводит и к тому, что их внутренняя энергия зависит от плотности. С этим свойством связан эффект Джоуля-Томпсона: изменение температуры газа при его адиабатическом расширении, напр. при протекании с малой постоянной скоростью через пористую перегородку (этот процесс называется дросселированием). Учет межмолекулярных взаимодействий и внутреннего строения молекул необходим при решении многих теоретических задач физики и физической химии. Молекул, которые можно было бы принимать как упругие шары, практически не бывает, и при расчете свойств реальных газов применяют другие молекулярные модели. Из них наиболее употребительны простые модели гармонического осциллятора и жесткого ротатора.
Реальные газы при незначительных плотностях имеют свойства, отличающиеся от свойств идеальных газов. Это различие свойств тем значительнее, чем выше плотность газа. Так, например, из уравнения Менделеева-Клайперона следует, что так называемый коэффициент сжимаемости для любого газа Zсж = pV/RT = 1. В действительности же коэффициент сжимаемости является переменной величиной, принимающей в зависимости от давления и температуры значения и большие, и меньшие единицы, и только при малых давлениях он равен единице. (см. рис 6.1)
Внутреннее строение молекул газа слабо влияет на их термические свойства (давление, температуру, плотность и связь между ними). Для этих свойств в первом приближении существенна только молекулярная масса реального газа. Напротив, его калорические свойства (теплоёмкость, энтропия и др.), а также его электрические и магнитные свойства существенно зависят от внутреннего строения молекул. Например, для расчёта (в первом приближении) теплоёмкости при постоянном объёме — Cv необходимо знать число внутренних степеней свободы молекулы (т. е. число возможных внутренних движений). В соответствии с законом равнораспределения классической статистической физики на каждую степень свободы молекулы газа (поступательную, колебательную, вращательную) приходится энергия, равная 1/2 · kT. Отсюда теплоёмкость 1 моля равна:
Отступление свойств реальных газов от свойств идеальных газов обнаруживается не только при изучении сжимаемости газов, но также при изучении калорических свойств газов, например их теплоемкостей. Теплоемкости Cv и Cp идеального газа не зависят от давления (или объема ) и являются функциями только температуры . В действительности теплоемкости всех газов зависят от давления или объема.
Для точного расчёта калорических свойств газа необходимо знать уровни энергии молекулы, сведения о которых в большинстве случаев получают из анализа спектров. Для большого числа веществ в состоянии идеального газа калорические свойства вычислены с высокой точностью и их значения представлены в виде таблиц до температур 10—22 тыс. градусов.
Электрические свойства газов связаны в первую очередь с возможностью ионизации молекул или атомов, т. е. с появлением в них электрически заряженных частиц (ионов и электронов). При отсутствии заряженных частиц газы являются хорошими диэлектриками. С ростом концентрации зарядов электропроводность увеличивается.
При температурах, начиная с нескольких тысяч градусов всякий газ частично ионизуется и превращается в плазму. Если концентрация зарядов в плазме невелика, то свойства её мало отличаются от свойств обычного газа.
По магнитным свойствам газы делятся на диамагнитные (к ним относятся, например, инертные газы, H2, N2, CO2, H2O) и парамагнитные (например, O2). Диамагнитны те газы, молекулы которых не имеют постоянного магнитного момента и приобретают его лишь под влиянием внешнего поля. Те же, у которых молекулы обладают постоянным магнитным моментом, во внешнем магнитном поле ведут себя как парамагнетики. Учёт межмолекулярного взаимодействия и внутреннего строения молекул необходим при решении многих проблем физики Г., например при исследовании влияния верхних разреженных слоев атмосферы на движение ракет и спутников.
Применение законов классичесской статистики с учетом квантовых закономерностей позволяет рассчитать по молекулярным данным термодинамические функции газа (энтропию, внутреннюю энергию, энергии Гельмгольца и Гиббса), константы химического равновесия газофазных реакций, теплоемкость и кинетические характеристики, знание которых требуется при проектировании многих технологических процессов. Так, теплоемкость идеального газа может быть рассчитана в классической теории, если известно число i степеней свободы молекулы. Вклад каждой из вра-щат. и постулат, степеней свободы молекулы в молярную теплоемкость Суд равен R/2, а каждой из колебательных степеней свободы-JR (т. наз. закон равнораспределения). Частица одноатомного газа обладает тремя степенями свободы, соотв. его теплоемкость составляет ЗЯ/2, что хорошо совпадает с экспериментальными данными. Молекула двухатомного газа обладает тремя поступательными, двумя вращательными и одной колебательной степенями свободы, и, согласно закону равнораспределения, Суд = 1R/2, однако это значение не совпадает с опытными данными даже при обычных температурах. Наблюдаемое расхождение, а также температурная зависимость теплоемкости газа объясняются квантовой теорией.
Внутренняя энергия реальных газов.
Внутренняя энергия реального газа складывается из кинетической энергии теплового движения его молекул и из потенциальной энергии межмолекулярного взаимодействия. Потенциальная энергия реального газа обусловлена только силами притяжения между молекулами. Наличие сил притяжения приводит к возникновению внутреннего давления на газ.
р΄=а/V2
Работа, которая затрачивается для преодоления сил притяжения, действующих между молекулами газа, или, иными словами, против внутреннего давления, как известно из механики, идёт на увеличение потенциальной энергии системы.
Т.е. dA=p΄Vm=dП, или dП=a/V2m*dVm, откуда П=-а/Vm.
Знак минус означает, что молекулярные силы, создающие внутреннее давление р΄, являются силами притяжения. Если газ расширяется без теплообмена с окружающей средой и не совершает внешней работы, то на основании первого начала термодинамики получим, что U1=U2. Следовательно, при адиабатическом расширении без совершения внешней работы внутренняя энергия газа не изменяется.
Использование выражения для внутренней энергии идеального газа позволяет записать формулу, для расчета внутренней энергии газа Ван-дер-Ваальса в виде:
Как следует из этого выражения, внутренняя энергия газа Ван-дер-Ваальса зависит не только от его температуры, как в случае с идеальным газом, но и от объема, занимаемого им. По этой причине, при осуществлении изотермических процессов в газе Ван-дер-Ваальса, будет изменяться его внутренняя энергия, а, следовательно, при таких процессах подведенная к газу теплота не будет равна совершенной им работе.
Если внутренняя энергия идеального газа определяется кинетической энергией его молекул, то для газа Ван-дер-Ваальса существенное значение имеет потенциальная энергия, обусловленная силами притяжения и отталкивания. Согласно первому началу термодинамики изменение внутренней энергии газа может быть осуществлено либо сообщением ему теплоты, либо совершением над ним механической работы. Если газ адиабатически расширяется, не совершая механической работы, его внутренняя энергия остается неизменной. Для идеального газа неизменность внутренней энергии эквивалентна постоянству температуры газа.
Термодинамические свойства реальных газов.
Как известно, уравнение состояния устанавливает функциональную связь между давлением p, объемом V, температурой T и числом молей n газа в состоянии равновесия. Эта связь может выражаться не только в форме уравнения, но также графически или в виде таблиц, которые часто используются, особенно для практических целей. Самым простым и известным уравнением состояния является уравнение состояния идеального газа:
pV = nRT, где R – универсальная газовая постоянная.
Реальные газы описываются уравнением состояния идеального газа только приближенно, и отклонения от идеального поведения становятся заметными при высоких давлениях и низких температурах, особенно когда газ близок к конденсации.
Так, для газов с низкой температурой сжижения (He, H2, Ne и даже N2, O2, Ar, CO, CH4) при давлениях до 50 атм отклонения не превышают 5%, а при давлениях до 10 атм – 2%. Легко конденсирующиеся газы (CO2, SO2, Cl2, CH3Cl) уже при 1 атм обнаруживают отклонения до 2 – 3%.
Одной из наглядных характеристик отклонений реальных газов от идеального поведения оказывается мольный объем газа Vm = V/n. Для идеального газа он равен 22,414 л при 1 атм и 273 K. Наиболее удобной мерой неидеальности является фактор сжимаемости Z = pVm/RT, поскольку для идеального газа Z = l при любых условиях.
Рис.1.1 Зависимость фактора сжимаемости некоторых газов от давления при 298 К.
На рис. 1.1 представлены факторы сжимаемости для некоторых реальных газов как функции давления при 298 К (для сравнения поведение идеального газа показано пунктиром). При высоких давлениях для всех газов Z > 1, т.е. их труднее сжать, чем идеальный газ, поскольку в этой области преобладают силы межмолекулярного отталкивания. Из рисунка видно, что при более низких давлениях для некоторых газов Z 1). Очевидно, что основными причинами отклонений свойств реальных газов от свойств идеального газа оказываются взаимное притяжение молекул и наличие у них собственного объема. Наиболее ярко межмолекулярное притяжение в реальных газах проявляется в их способности к конденсации – переходу в жидкое состояние.
При понижении температуры или повышении давления наблюдаются отклонения от законов идеального газа. Когда T и P достигают некоторых определенных значений, то происходит конденсация газа, т.е. газ переходит в жидкость. Это явление уже никак не следует из уравнения состояния идеального газа. Рассмотрим его более подробно.
Рассмотрим, что происходит, когда образец газа в состоянии, отмеченном точкой А на рис. 1.3, сжимается при постоянной температуре.
Рис 1.3. Экспериментальные изотермы для СО2
Вблизи точки А давление возрастает приблизительно по закону Бойля. Заметные отклонения от закона Бойля начинают наблюдаться, когда объем становится соизмеримым со значением, указанным точкой В. В точке С сходство с идеальным поведением полностью теряется, так как оказывается, что дальнейшее уменьшение объема не вызывает роста давления; это показано горизонтальной линией CDE. Исследование содержания сосуда показывает, что сразу за точкой С появляется жидкость, и можно наблюдать две фазы, разделенные резко обозначенной границей – поверхностью раздела. Поскольку при уменьшении объема газ конденсируется, он не оказывает сопротивления дальнейшему движению поршня. Давление, соответствующее линии CDE, когда жидкость и пар находятся в равновесии, называется давлением пара жидкости при температуре опыта.
В точке Е весь образец представляет собой жидкость, и дальнейшее уменьшение объема образца требует значительного давления, поскольку жидкости по сравнению с газами очень трудно сжимаются, что проявляется в резком подъеме кривой слева от точки Е.
Изотерма при температуре Tк играет особую роль в теории состояния вещества. Изотерма, соответствующая температуре ниже Tc, ведет себя так, как уже описано: при определенном давлении газ конденсируется в жидкость, которую можно различать по наличию поверхности раздела. Если же сжатие осуществлять при Tc, то поверхность, разделяющая две фазы, не появляется, а точка конденсации и точка полного перехода в жидкость сливаются в одну критическую точку газа. При температуре выше Tc газ невозможно обратить в жидкость никаким сжатием. Температура, давление и мольный объем в критической точке называются критической температурой Tc, критическим давлением pc и критическим мольным объемом Vc вещества. Собирательно параметры pк, Vк, и Tк называются критическими константами данного газа.
При T > Tк образец представляет собой фазу, полностью занимающую объем содержащего ее сосуда, т. е. по определению является газом. Однако плотность этой фазы может быть значительно большей, чем это типично для газов, поэтому обычно предпочитают название «сверхкритический флюид» (supercritical fluid).
В критической точке изотермический коэффициент сжимаемости равен бесконечности, поскольку = 0. Поэтому вблизи критической точки сжимаемость вещества так велика, что ускорение силы тяжести приводит к значительным различиям плотности в верхней и нижней частях сосуда, достигающим 10% в столбике вещества высотой всего несколько сантиметров. Это затрудняет определение плотностей (удельных объемов) и, соответственно, изотерм p – V вблизи критической точки. В то же время критическую температуру можно определить весьма точно как такую температуру, при которой поверхность, разделяющая газообразную и жидкую фазы, исчезает при нагревании и вновь появляется при охлаждении. Зная критическую температуру, можно определить критическую плотность (и, соответственно, критический мольный объем), пользуясь эмпирическим правилом прямолинейного диаметра (правило Кальете-Матиаса), согласно которому средняя плотность жидкости и насыщенного пара является линейной функцией температуры:
, где A и B – постоянные для данного вещества величины. Экстраполируя прямую средней плотности до критической температуры, можно определить критическую плотность.
Высокая сжимаемость вещества вблизи критической точки приводит к росту спонтанных флуктуаций плотности, которые сопровождаются аномальным рассеянием света. Это явление называется критической опалесценцией.
Поведение газов, которые близки к конденсации, не описывается уравнением состояния идеального газа. Однако это уравнение можно усовершенствовать так, чтобы оно приближенно описывало не только свойства газа, но и свойства жидкости. Предпринималось множество попыток для учета отклонений свойств реальных газов от свойств идеального газа путем введения различных поправок в уравнение состояния идеального газа. Наибольшее распространение вследствие простоты и физической наглядности получило уравнение Иоханнеса Дидерика Ван-дер-Ваальса (1837 — 1923).
Первая поправка в уравнении состояния идеального газа рассматривает собственный объем, занимаемый молекулами реального газа. В уравнении Дюпре (1864): p(V – νb) = νRT, где постоянная b учитывает собственный мольный объем молекул.
При понижении температуры межмолекулярное взаимодействие в реальных газах приводит к конденсации (образование жидкости). Межмолекулярное притяжение эквивалентно существованию в газе некоторого внутреннего давления (иногда его называют статическим давлением). Изначально величина была учтена в общей форме в уравнении Гирна (1865): (p + π ) (V – νb) = νRT.
Ван-дер-Ваальс в 1873 г. дал функциональную интерпретацию внутреннего давления. Согласно модели Ван-дер-Ваальса, силы притяжения между молекулами (силы Ван-дер-Ваальса) обратно пропорциональны шестой степени расстояния между ними, или второй степени объема, занимаемого газом. Считается также, что силы притяжения суммируются с внешним давлением. С учетом этих соображений уравнение состояния идеального газа преобразуется в уравнение Ван-дер-Ваальса:
Перепишем это уравнение так, чтобы выразить объем:
Это уравнение содержит объем в третьей степени и, следовательно, имеет или три действительных корня, или один действительный и два мнимых. При высоких температурах оно имеет один действительный корень, и по мере повышения температуры кривые, вычисленные по уравнению Ван-дер-Ваальса, приближаются к гиперболам, соответствующим уравнению состояния идеального газа.
Рис.1.4 Изотермы Ван-дер-Ваальса для СО2
На рис. 1.4 приведены изотермы, вычисленные по уравнению Ван-дер-Ваальса для диоксида углерода. Из рисунка видно, что при температурах ниже критической (31,04 °С) вместо горизонтальных прямых, соответствующих равновесию жидкости и пара, получаются волнообразные кривые 12345 с тремя действительными корнями, из которых только два, 1 и 5, физически осуществимы. Третий корень (точка 3) физически не реален, поскольку находится на участке кривой 234, противоречащем условию стабильности термодинамической системы. Состояния на участках 12 и 54, которые соответствуют переохлажденному пару и перегретой жидкости, соответственно, являются неустойчивыми (метастабильными) и могут быть лишь частично реализуемы в специальных условиях. Так, осторожно сжимая пар выше точки 1 (рис. 1.4), можно подняться по кривой 12. Для этого необходимо отсутствие в паре центров конденсации, и в первую очередь пыли. В этом случае пар оказывается в пересыщенном, т.е. переохлажденном состоянии. И наоборот, образованию капелек жидкости в таком паре способствуют, например, попадающие в него ионы. Это свойство пересыщенного пара используется в известной камере Вильсона (1912), применяемой для регистрации заряженных частиц. Движущаяся заряженная частица, попадая в камеру, содержащую пересыщенный пар, и соударяясь с молекулами, образует на своем пути ионы, создающие туманный след – трек, который фиксируется фотографически.
Согласно правилу Максвелла (the Maxwell construction), которое имеет теоретическое обоснование, для того, чтобы расчетная кривая соответствовала экспериментальной равновесной изотерме, нужно вместо кривой 12345 провести горизонтальную прямую 15 так, чтобы площади 1231 и 3453 были равны. Тогда ордината прямой 15 будет равна давлению насыщенного пара, а абсциссы точек 1 и 5 – мольным объемам пара и жидкости при данной температуре.
По мере повышения температуры все три корня сближаются, и при критической температуре Tc все три корня становятся равными. В критической точке изотерма Ван-дер-Ваальса имеет точку перегиба с горизонтальной касательной , то есть и .
Совместное решение этих уравнений дает вывод критических параметров. Соответственно, согласно уравнению Ван-дер-Ваальса, критический фактор сжимаемости Z для всех газов должен быть равен:
Принципиальное значение уравнения Ван-дер-Ваальса определяется следующими обстоятельствами:
1) уравнение было получено из модельных представлений о свойствах реальных газов и жидкостей, а не явилось результатом эмпирического подбора функции f(p,V,T), описывающей свойства реальных газов;
2) уравнение долго рассматривалось как некоторый общий вид уравнения состояния реальных газов, на основе которого было построено много других уравнений состояния (см. ниже);
3) с помощью уравнения Ван-дер-Ваальса впервые удалось описать явление перехода газа в жидкость и проанализировать критические явления. В этом отношении уравнение Ван-дер-Ваальса имеет преимущество даже перед более точными уравнениями в вириальной форме (см. 1.1, 1.2).
Причиной недостаточной точности уравнения Ван-дер-Ваальс считал ассоциацию молекул в газовой фазе, которую не удается описать, учитывая зависимость параметров a и b от объема и температуры, без использования дополнительных постоянных. После 1873 г. сам Ван-дер-Ваальс предложил еще шесть вариантов своего уравнения, последнее из которых относится к 1911 г. и содержит пять эмпирических постоянных. Две модификации уравнения предложил Клаузиус, и обе они связаны с усложнением вида постоянной b. Больцман получил три уравнения этого типа, изменяя выражения для постоянной a. Всего известно более сотни подобных уравнений, отличающихся числом эмпирических постоянных, степенью точности и областью применимости. Выяснилось, что ни одно из уравнений состояния, содержащих менее 5 индивидуальных постоянных, не оказалось достаточно точным для описания реальных газов в широком диапазоне p, V, T, и все эти уравнения оказались непригодными в области конденсации газов. Из простых уравнений с двумя индивидуальными параметрами неплохие результаты дают уравнения Дитеричи и Бертло.
Вириальное уравнение состояния.
Поведение реального газа можно описать с высокой точностью с помощью вириального уравнения (или уравнения с вириальными коэффициентами). Идея состоит в отказе от минимального числа параметров и использовании бесконечных рядов — разложений по степеням 1/V:
Коэффициенты B2, B3, . (которые зависят от температуры и природы рассматриваемого газа, но не зависят от плотности и давления) называются соответственно вторым, третьим, . вириальными коэффициентами. Первый вириальный коэффициент равен 1. Второй вириальный коэффициент обычно более важен, чем последующие, поскольку для большинства случаев B2 /Vm >> B3 /V2m >>. .
Уравнение состояния в виде бесконечного ряда было предложено Тиссеном в 1885 г. Однако основное развитие вириальное уравнение получило в 1901 г. в работе Камерлинг-Оннеса, который рассмотрел несколько вариантов этого уравнения и предложил называть его коэффициенты вириальными.
Если подходить в вириальному уравнению только как к эмпирическому уравнению состояния, то оно имеет ряд недостатков. Например, как показывают экспериментальные данные, сходимость ряда не очень хорошая, особенно в области высокой плотности. Кроме того, при высоких плотностях для удовлетворительного описания экспериментальных данных необходимо использовать большое число членов ряда, а для этого нужно экспериментально определять большое число вириальных коэффициентов. Более того, часто тот же набор экспериментальных данных можно более точно описать с помощью других эмпирических уравнений с меньшим числом параметров. Однако исключительная важность вириального уравнения состояния заключается в том, что это единственное из известных уравнений состояния, имеющее строгую теоретическую основу. Как будет показано в главе 2, каждый вириальный коэффициент можно выразить через силы межмолекулярного взаимодействия. Так, второй вириальный коэффициент отражает парные взаимодействия, третий – тройные и т.д. Таким образом, вириальное уравнение состояния позволяет объяснить свойства газа с позиции межмолекулярных взаимодействий.
Для некоторых целей вириальное уравнение удобнее записать в виде разложения по степеням p: pVm = RT (1 + B2’p + B3’p2 + . )
Таким образом, вириальное уравнение является примером того, когда простое выражение (в данном случае pVm = RT) представляет собой только первый член ряда разложения по степеням переменной (в данном случае p или Vm).
Рис.1.5. Зависимость второго вириального коэффициента некоторых газов от температуры.
Уравнения состояния реального газа
1.1. Уравнения состояния реального газа
Модель идеального газа хорошо описывает свойства газообразного состояния вещества при средних и высоких температурах (от комнатной и выше) и небольших давлениях (около атмосферного). Расчет свойств газов в широком интервале экспериментальных условий требует использования уравнения состояния реального газа[1].
Реальным газом называется газ, между молекулами которого существуют заметные силы межмолекулярного взаимодействия. Оно имеет электромагнитную и квантовую природу и осуществляется посредством сил межмолекулярного притяжения и отталкивания.
Силы притяжения, проявляющиеся на расстояниях r между центрами молекул порядка 10 -7 см, называются ван-дер-ваальсовыми силами. Они убывают с расстоянием r –7 , что соответствует изменению потенциальной энергии по закону r –6 .
Различают три вида ван-дер-ваальсовых сил [7]:
Ориентационные силы между двумя молекулами, обладающими постоянными дипольными моментами. Они стремятся расположить молекулы упорядоченно так, чтобы векторы дипольных моментов ориентировались вдоль одной прямой. Этому препятствует тепловое движение молекул.
Индукционные силы, возникающие между молекулами, обладающими высокой поляризуемостью. Если молекулы достаточно сближены, то под действием электрического поля одной из них в другой возникает индуцированный дипольный момент.
Дисперсионные силы возникают в результате возбуждения колебаний электронов в молекуле (атоме) под влиянием колебаний электронов в другой молекуле (атоме). Колебания электронов соседних молекул происходят в одинаковой фазе и приводят к притяжению двух молекул (атомов). Величина дисперсионных сил определяется нулевой энергией молекул (атомов), если их колебания можно рассматривать как колебания линейных гармонических осцилляторов.
Полная потенциальная энергия ван-дер-ваальсовых сил описывается суммой:
Для полярных молекул основную роль играют ориентационные силы притяжения, для остальных молекул – дисперсионные силы. Энергия ван-дер-ваальсового притяжения составляет (0,1 – 1) ккал/моль [7]. В большинстве случаев ван-дер-ваальсовы силы притяжения перекрываются значительно превосходящими их химическими валентными силами притяжения с энергиями порядка (10 – 100) ккал/моль.
Согласно упрощенной модели ван-дер-ваальсовых сил, молекулы газа – абсолютно упругие шары – притягиваются с силами, достигающими наибольшего значения при непосредственном их соприкосновении. Силы отталкивания проявляют себя на значительно меньших расстояниях.
Для описания свойств реальных газов применяют различные уравнения состояния, отличные от уравнения Клапейрона-Менделеева. Наиболее удобны двухпараметрические уравнения, разрешимые относительно давления и содержащие объем в третьей степени (кубические уравнения состояния). Первое такое уравнение было предложено Ван-дер-Ваальсом в 1873 г.
Уравнение Ван-дер-Ваальса состояния реального газа имеет следующий вид [7]:
,
где V0 – объем 1 моля газа, а – внутреннее давление, обусловленное силами притяжения между молекулами, b – поправка за собственный объем молекул, учитывающая действие сил отталкивания между молекулами и равная учетверенному объему молекул в 1 моле газа:
,
.
Здесь NA – число Авогадро, d – диаметр молекулы, U(r) – потенциальная энергия притяжения двух молекул.
Уравнение состояния Бертло (1900г.):
.
Здесь а и b связаны с параметрами критического состояния (в критической точке) соотношениями [8]:
.
Уравнение состояния Вукаловича и Новикова [7]:
.
Здесь B1, B2 и т.д. – так называемые вириальные коэффициенты весьма сложного вида. Их вычисление производится с учетом ассоциации молекул – объединения под влиянием ван-дер-ваальсовых сил притяжения.
Уравнение состояния Майера [7]:
,
где: di=dqi1*. dqin.
Здесь Uпij – взаимная потенциальная энергия i-й и j-й молекул, взаимодействующих по закону центральных сил, qi1. qin – обобщенные координаты i-той молекулы, обладающей n степенями свободы.
Уравнение Камерлинг-Оннеса (1901) [8]:
где , .
Уравнение Редлиха-Квонга (1949 г.) [8]:
Здесь 0,42748·R 2 ·T 2,5 k/Pk, b = 0,08664·R·Tk/Pk. Уравнение Редлиха-Квонга считается наилучшим двухконстантным уравнением. При его выводе авторы не руководствовались какими-то определенными теоретическими обоснованиями [8]. Это уравнение следует рассматривать как произвольную, но удачную эмпирическую модификацию предшествующих уравнений состояния.
Уравнение Мартина (1967 г.) [8]:
,
где 27·R 2 ·T 2 k/(64Pk), b = R·Tk/(8Pk).
Реальные газы
Автор работы: Пользователь скрыл имя, 12 Декабря 2013 в 19:09, реферат
Краткое описание
Газы (французское gaz; название предложено голланским учёным Я. Б. Гельмонтом), агрегатное состояние вещества, в котором его частицы не связаны или весьма слабо связаны силами взаимодействия и движутся свободно, заполняя весь предоставленный им объём. Вещество в газообразном состоянии широко распространено в природе. Газы образуют атмосферу Земли, в значительных количествах содержатся в твёрдых земных породах, растворены в воде океанов, морей и рек.
Содержание
Введение. 1.Понятие реального газа. 2.Внутренняя энергия реального газа. 3.Уравнение Ван-дер-Ваальса. 4.Изотермы Ван-дер-Ваальса. 5.Фазовые переходы первого и второго рода. 6.Третье начало термодинамики(теорема Нернста). Заключение. Список литературы.
Вложенные файлы: 1 файл
Реальные газа.docx
Понятие реального газа.
Внутренняя энергия реального газа.
Уравнение Ван-дер-Ваальса.
Изотермы Ван-дер-Ваальса.
Фазовые переходы первого и второго рода.
Третье начало термодинамики(теорема Нернста).
Газы (французское gaz; название предложено голланским учёным Я. Б. Гельмонтом), агрегатное состояние вещества, в котором его частицы не связаны или весьма слабо связаны силами взаимодействия и движутся свободно, заполняя весь предоставленный им объём. Вещество в газообразном состоянии широко распространено в природе. Газы образуют атмосферу Земли, в значительных количествах содержатся в твёрдых земных породах, растворены в воде океанов, морей и рек. Солнце, звёзды, облака межзвёздного вещества состоят из газов — нейтральных или ионизованных (плазмы). Встречающиеся в природных условиях газы представляют собой, как правило, смеси химически индивидуальных газов. Газы обладают рядом характерных свойств. Они полностью заполняют сосуд, в котором находятся, и принимают его форму. В отличие от твёрдых тел и жидкостей, объём газа существенно зависит от давления и температуры. Коэффициент объёмного расширения газа в обычных условиях (0-100°С) на два порядка выше, чем у жидкостей, и составляет в среднем 0,003663 град-1.
Реальный газ-это модель идеального газа, используемая в молекулярно-кинетической теории газов, позволяющая описывать поведение разрежённых реальных газов при достаточно высоких температурах и низких давлениях. При выводе уравнения состояния идеального газа размерами молекул и их взаимодействием друг с другом пренебрегают. Повышение давления приводит к уменьшению среднего расстояния между молекулами, поэтому необходимо учитывать объём молекул и взаимодействие между ними. При высоких давлениях и низких температурах указанная модель идеального газа непригодна. При рассмотрении реальных газов – газов, свойства которых зависят от взаимодействия молекул, надо учитывать силы межмолекулярного взаимодействия. Они проявляются на расстояниях ≤10-9 м. и быстро убывают при увеличении расстояния между молекулами. Такие силы называются короткодействующими. В ХХ в., по мере развития и представлений о строении атома и квантовой механики, было выяснено, что между молекулами вещества одновременно действуют силы притяжения и силы отталкивания. Силы отталкивания считаются положительными, а силы взаимного притяжения – отрицательными.
Внутренняя энергия реального газа складывается из кинетической энергии теплового движения его молекул и из потенциальной энергии межмолекулярного взаимодействия. Потенциальная энергия реального газа обусловлена только силами притяжения между молекулами. Наличие сил притяжения приводит к возникновению внутреннего давления на газ. р΄=а/V2
Работа, которая затрачивается для преодоления сил притяжения, действующих между молекулами газа, или, иными словами, против внутреннего давления, как известно из механики, идёт на увеличение потенциальной энергии системы.
Т.е. dA=p΄Vm=dП, или dП=a/V2m*dVm, откуда П=-а/Vm.
Знак минус означает, что молекулярные силы, создающие внутреннее давление р΄, являются силами притяжения. Учитывая оба слагаемых, получим, что внутренняя энергия моля реального газа Um=CVT-a/Vm растёт с повышением температуры и увеличением объёма. Если газ расширяется без теплообмена с окружающей средой и не совершает внешней работы, то на основании первого начала термодинамики получим, что U1=U2. Следовательно, при адиабатическом расширении без совершения внешней работы внутренняя энергия газа не изменяется.
Учёт собственного объёма молекул и сил межмолекулярного взаимодействия привёл голландского физика И. Ван-дер-Ваальса (1837-1923) к выводу уравнения состояния реального газа. Ван-дер-Ваальсом в уравнение Клапейрона- Менделеева введены две поправки.
1. Учёт собственного объёма молекул. Наличие сил отталкивания, которые противодействуют проникновению в занятый молекулой объём других молекул, сводится к тому, что фактический свободный объём, в котором могут двигаться молекулы реального газа, будет не Vm, а Vm-b,
где b- объём, занимаемый самими молекулами. Объём b равен утверждённому собственному объёму молекул. Если, например, в сосуде находятся две молекулы, то центр любой из них не может приблизиться к центру другой молекулы на расстояние меньше d, это означает, что для центров обеих молекул оказывается недоступным объём сферы радиусом d, объём, равный восьми объёмам молекулы, а в расчёте на одну молекулу – учетверённый объём молекулы.
2. Учёт притяжения молекул. Действие сил притяжения между молекулами реального газа приводит к появлению дополнительного давления на газ, называемого внутренним давлением. По вычислениям Ван-дер-Ваальса, внутреннее давление обратно квадрату объёма газа, т.е. p΄=a/V2 , где а – постоянная Ван-дер-Ваальса, характеризующая силы межмолекулярного притяжения, Vm – молярный объём. Вводя эти поправки – получим уравнение Ван-дер-Ваальса для моля газа (уравнение состояния идеальных газов): (p+a/V2m) (Vm-b)=RT. При выводе уравнения Ван-дер-Ваальса сделан целый ряд упрощений, поэтому оно также весьма приближённое, хотя и лучше согласуется с опытом, чем уравнение состояния идеального газа. При малых давлениях и высоких температурах объём Vm становится большим, поэтому b Тк) изотерма реального газа отличается от изотермы идеального газа только некоторым искажением её формы, оставаясь монотонно спадающей кривой; при некоторой температуре, на изотерме имеется лишь одна точка перегиба; при низких температурах (Т Тк) изотерма реального газа отличается от изотермы идеального газа только некоторым искажением её формы, оставаясь монотонно спадающей кривой; при некоторой температуре, на изотерме имеется лишь одна точка перегиба; при низких температурах (Т рассматривают, пользуясь моделью идеального кристалла, т. е. предполагая регулярное расположение всех атомов. Парообразную же ветвь получают, используя модель идеального газа, предполагающую полный беспорядок в системе. Зависимости, полученные для различных моделей, накладывают друг на друга и исследуют, какая из возможностей реализуется в данных условиях. Получить описание фазового перехода первого рода, одновременно учитывая все состояния системы, до настоящего времени не удается из-за огромных математических трудностей. Фазовые переходы, не связанные с поглощением или выделением теплоты и изменением объёма, называются фазовыми переходами второго рода. Эти переходы характеризуются постоянством объёма и энтропии, но скачкообразным изменением теплоёмкости. Общая трактовка фазовых переходов второго рода предложена советским учёным Л.Д.Ландау (1908-1968). Согласно этой трактовке, фазовые переходы второго рода связаны с изменением симметрии: выше точки перехода система, как правило, обладает более высокой симметрией, чем ниже точки перехода. Примерами фазовых переходов второго рода являются: переход ферромагнитных веществ (железа, никеля) при определённых давлении и температуре в парамагнитное состояние; переход металлов и некоторых сплавов при температуре, близкой к 0К, в сверхпроводящее состояние, характеризуемое скачкообразным уменьшением электрического сопротивления до нуля; превращение обыкновенного жидкого гелия при Т=2,9К в другую жидкую модификацию, обладающую свойствами сверхтекучести.
Третье начало термодинамики(теорема Нернста).
Третье начало термодинамики было сформулировано в 1906 году немецким физиком и химиком Вольтером Фридрихом Германом Нернстом (1864 — 1941) эмпирическим путем на основе обобщения экспериментальных данных и получило название теоремы Нернста: При стремлении температуры любой равновесной термодинамической системы к абсолютному нулю ее энтропия стремится к некоторой универсальной постоянной величине, значение которой не зависит от каких-либо термодинамических параметров системы и может быть принято равной нулю:
Из утверждения теоремы Нернста о независимости значения энтропии равновесной системы при абсолютном нуле температуры от ее термодинамических параметров следует также выражение:
где — любой термодинамический параметр системы, например, объем, давление и т.д. Здесь нижний индекс за скобками обозначает дифференцирование при постоянном значение величины . Теорема Нернста применима только для систем, находящихся в состоянии термодинамического равновесия и не справедлива для неравновесных систем. В частности, при стремлении температуры аморфного тела, например, стекла, к абсолютному нулю, его энтропия не стремится к некоторому определенному постоянному значению. В зависимости от того, как осуществляется процесс охлаждения, энтропия аморфного тела при стремлении к абсолютному нулю будет различной. Это связано с тем, что для аморфных тел, которые находятся в неравновесном (метастабильном) состоянии, процесс охлаждения может происходить быстрее, чем переход их в равновесное (кристаллическое) состояние. Из третьего начала термодинамики непосредственно следует недостижимость температуры равной абсолютному нулю. Действительно, для того, чтобы практически осуществить охлаждение термодинамической системы до абсолютного нуля температуры, необходимо чередовать изотермическое сжатие и адиабатическое расширение. При первом процессе происходит отвод теплоты, а при втором — уменьшение температуры системы энтропии. Другим следствием третьего начала термодинамики является невозможность использования уравнения Клапейрона-Менделеева для описания идеального газа при температурах, близких к абсолютному нулю. Так как для идеального газа на основании первого начала термодинамики можно записать:
то определение энтропии с помощью интеграла дает:
где — произвольная постоянная интегрирования. Здесь из соображений размерности введены величины и , которые можно считать равными единице в системе СИ: К и м3. Таким образом, при энтропия, вычисленная по формуле (4), не принимает нулевого значения, а стремится к минус бесконечности. А это противоречит третьему началу термодинамики, что делает невозможным применение уравнения Клапейрона-Менделеева для описания газа при температурах, близких к абсолютному нулю. Состояние газа при называется вырожденным состоянием и для его описания требуется применение законов, следующих из уравнений квантовой статистики.





 равен бесконечности, поскольку
равен бесконечности, поскольку  = 0. Поэтому вблизи критической точки сжимаемость вещества так велика, что ускорение силы тяжести приводит к значительным различиям плотности в верхней и нижней частях сосуда, достигающим 10% в столбике вещества высотой всего несколько сантиметров. Это затрудняет определение плотностей (удельных объемов) и, соответственно, изотерм p – V вблизи критической точки. В то же время критическую температуру можно определить весьма точно как такую температуру, при которой поверхность, разделяющая газообразную и жидкую фазы, исчезает при нагревании и вновь появляется при охлаждении. Зная критическую температуру, можно определить критическую плотность (и, соответственно, критический мольный объем), пользуясь эмпирическим правилом прямолинейного диаметра (правило Кальете-Матиаса), согласно которому средняя плотность жидкости и насыщенного пара является линейной функцией температуры:
= 0. Поэтому вблизи критической точки сжимаемость вещества так велика, что ускорение силы тяжести приводит к значительным различиям плотности в верхней и нижней частях сосуда, достигающим 10% в столбике вещества высотой всего несколько сантиметров. Это затрудняет определение плотностей (удельных объемов) и, соответственно, изотерм p – V вблизи критической точки. В то же время критическую температуру можно определить весьма точно как такую температуру, при которой поверхность, разделяющая газообразную и жидкую фазы, исчезает при нагревании и вновь появляется при охлаждении. Зная критическую температуру, можно определить критическую плотность (и, соответственно, критический мольный объем), пользуясь эмпирическим правилом прямолинейного диаметра (правило Кальете-Матиаса), согласно которому средняя плотность жидкости и насыщенного пара является линейной функцией температуры:



 с горизонтальной касательной
с горизонтальной касательной  , то есть
, то есть  и
и  .
.


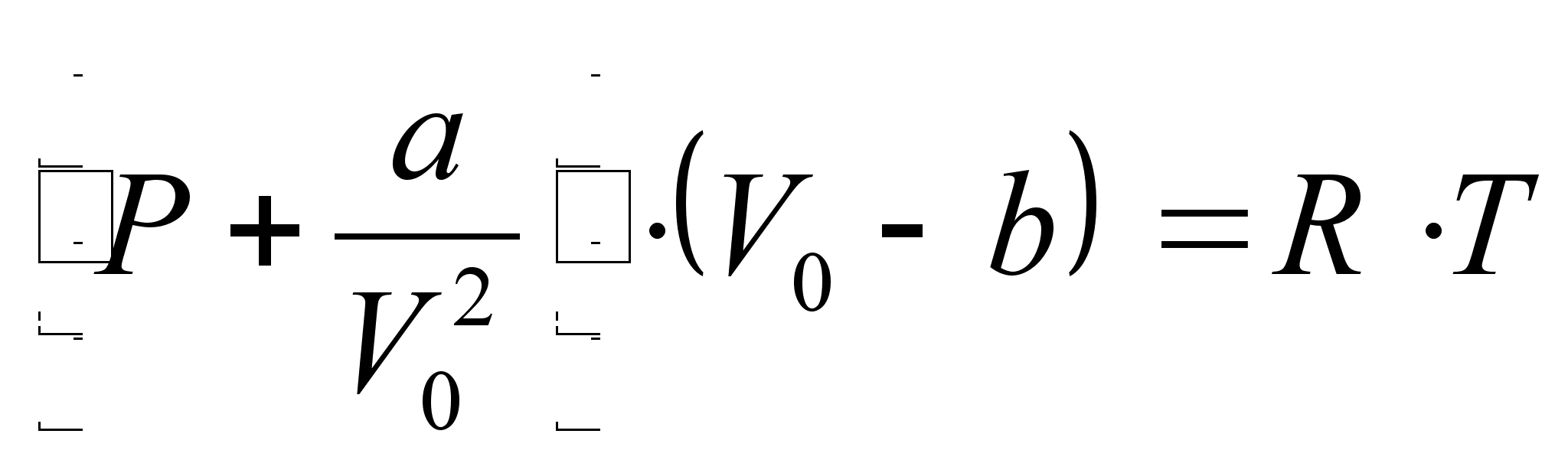 ,
,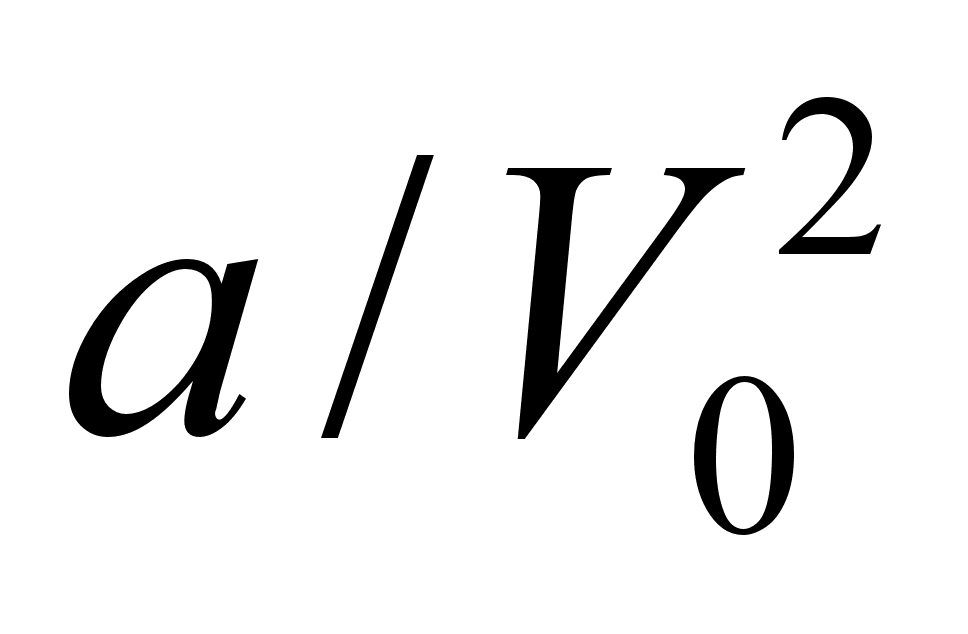 – внутреннее давление, обусловленное силами притяжения между молекулами, b – поправка за собственный объем молекул, учитывающая действие сил отталкивания между молекулами и равная учетверенному объему молекул в 1 моле газа:
– внутреннее давление, обусловленное силами притяжения между молекулами, b – поправка за собственный объем молекул, учитывающая действие сил отталкивания между молекулами и равная учетверенному объему молекул в 1 моле газа: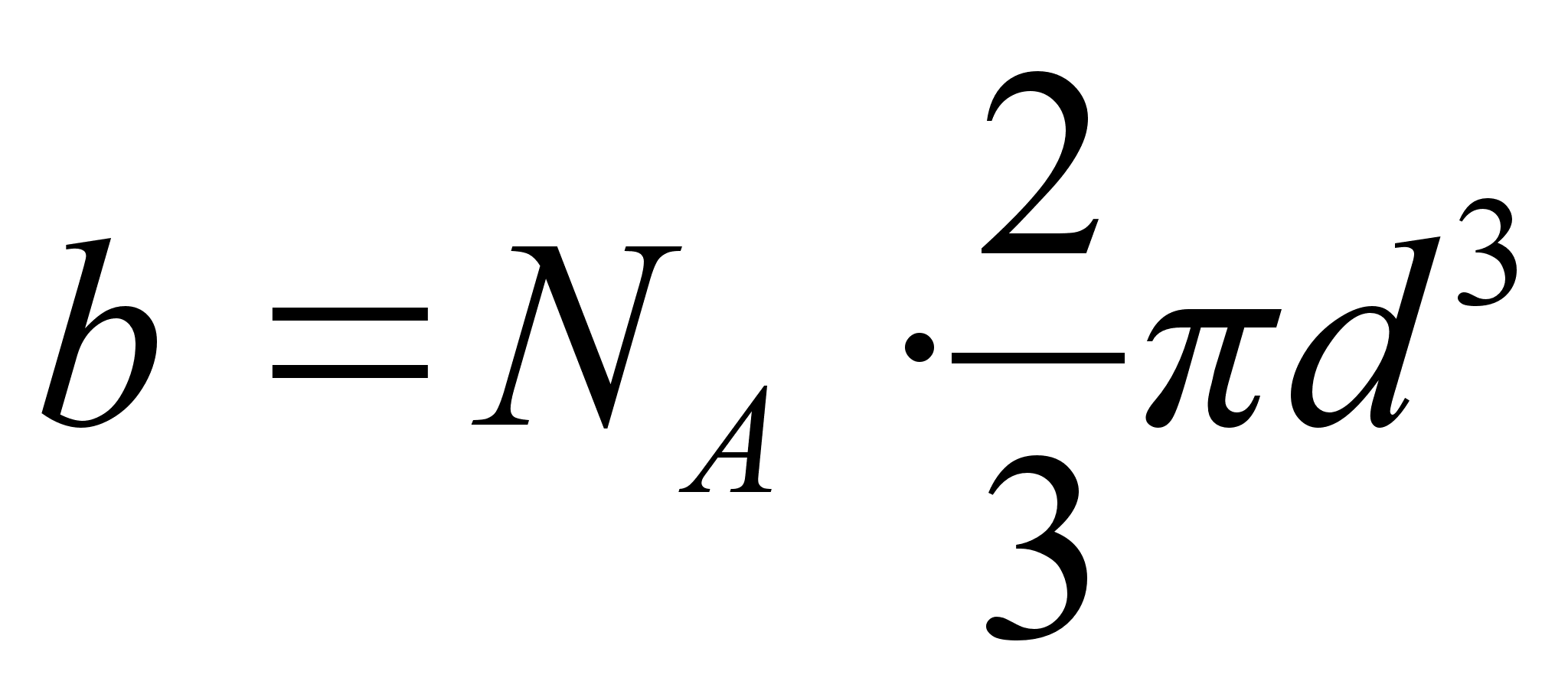 ,
,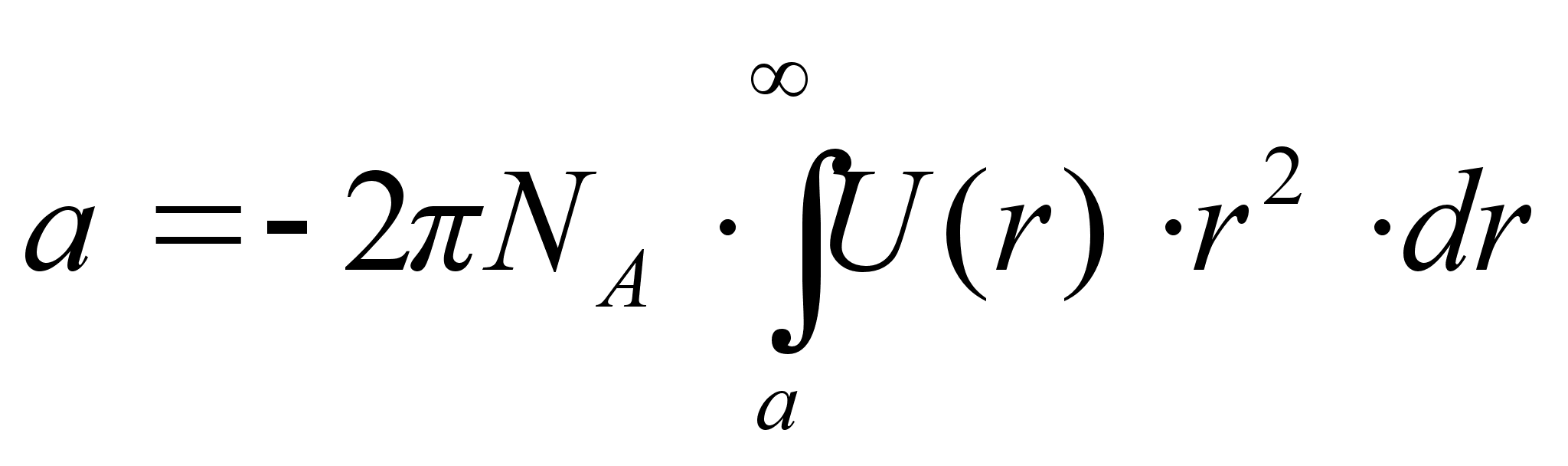 .
. .
.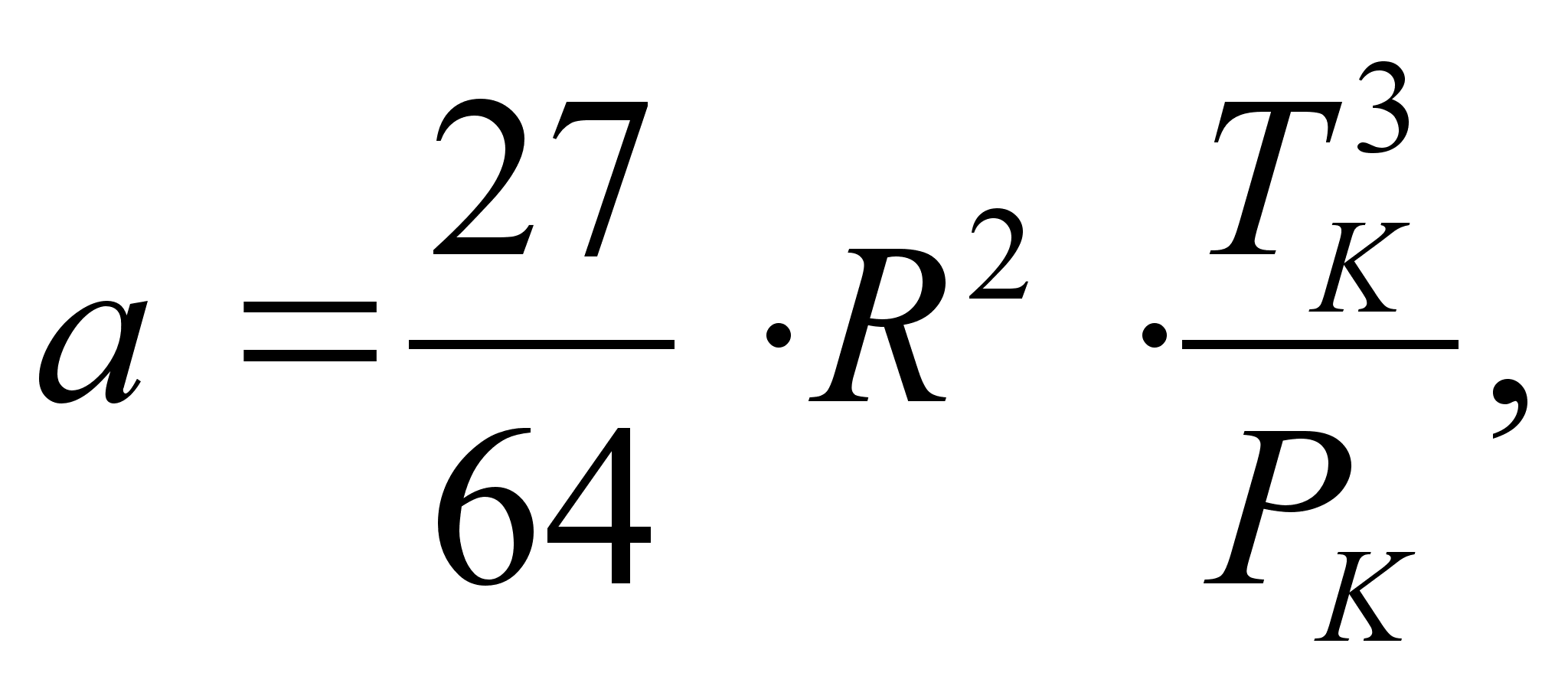
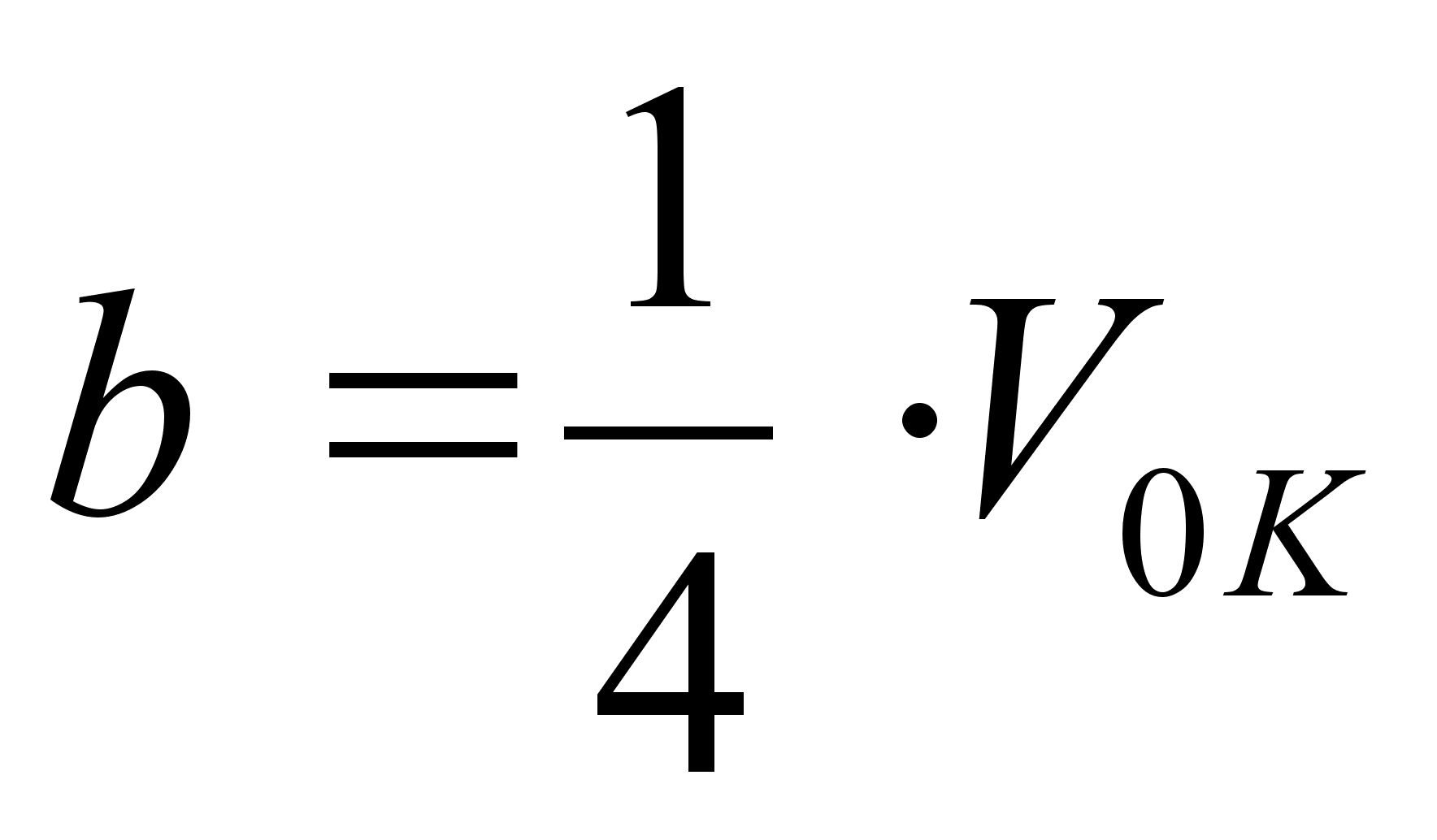 .
.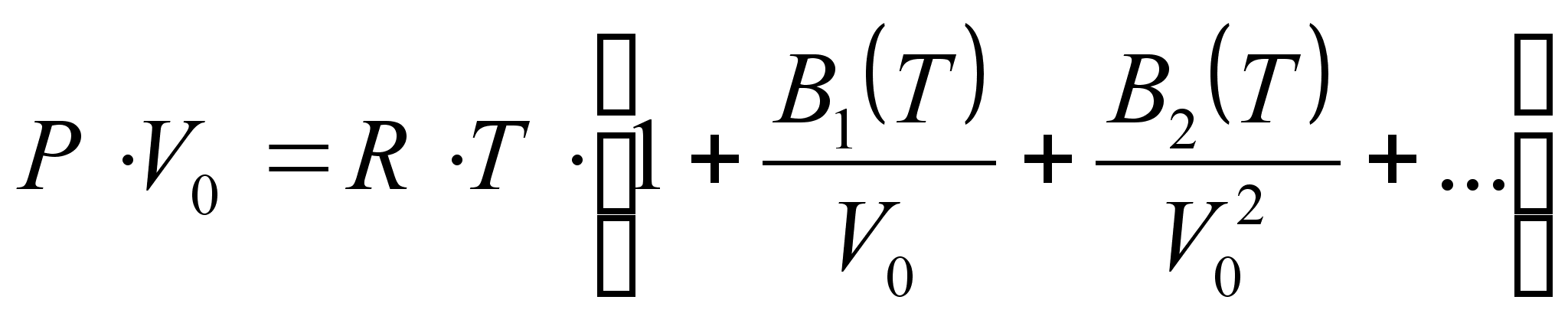 .
. ,
,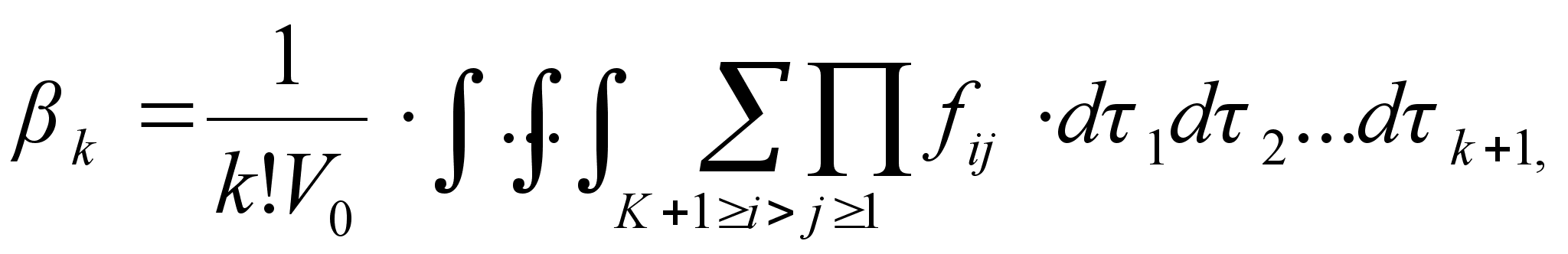
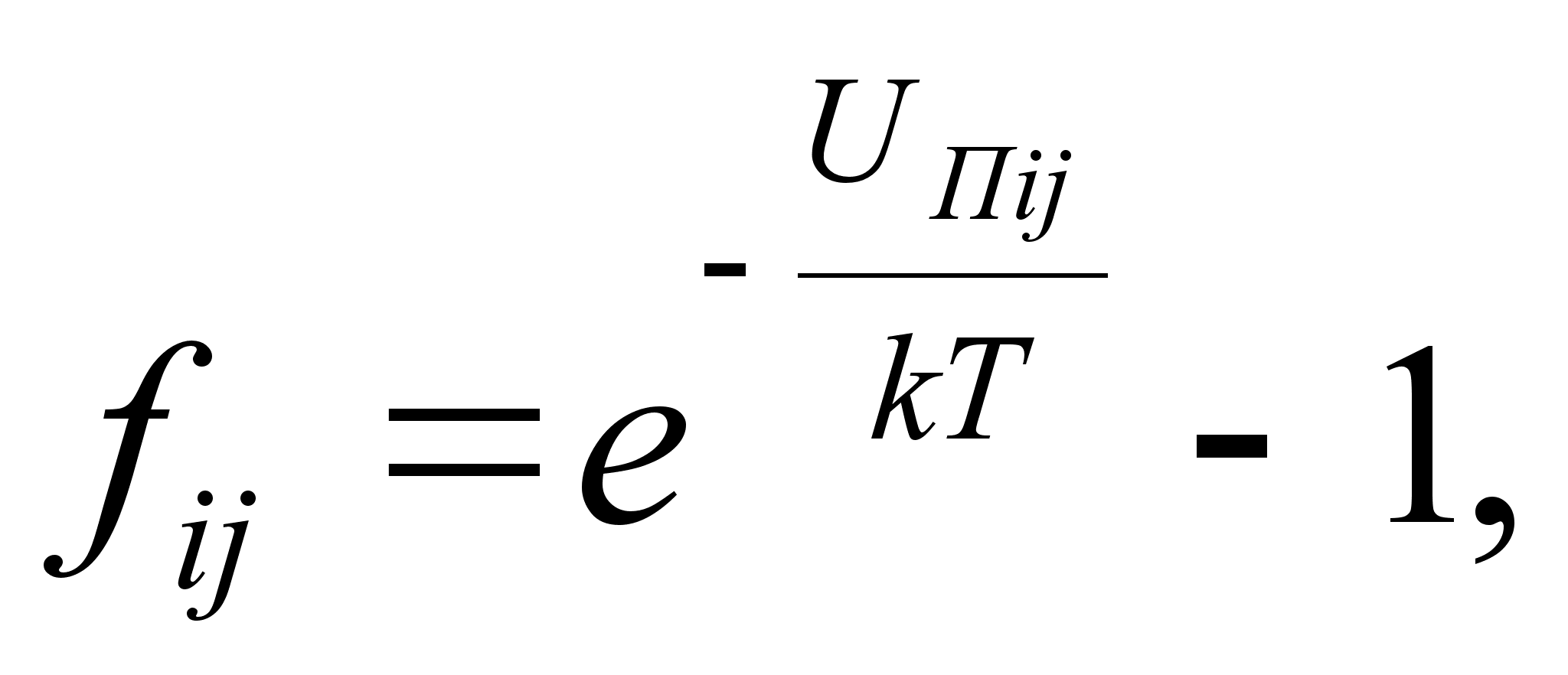 di=dqi1*. dqin.
di=dqi1*. dqin.
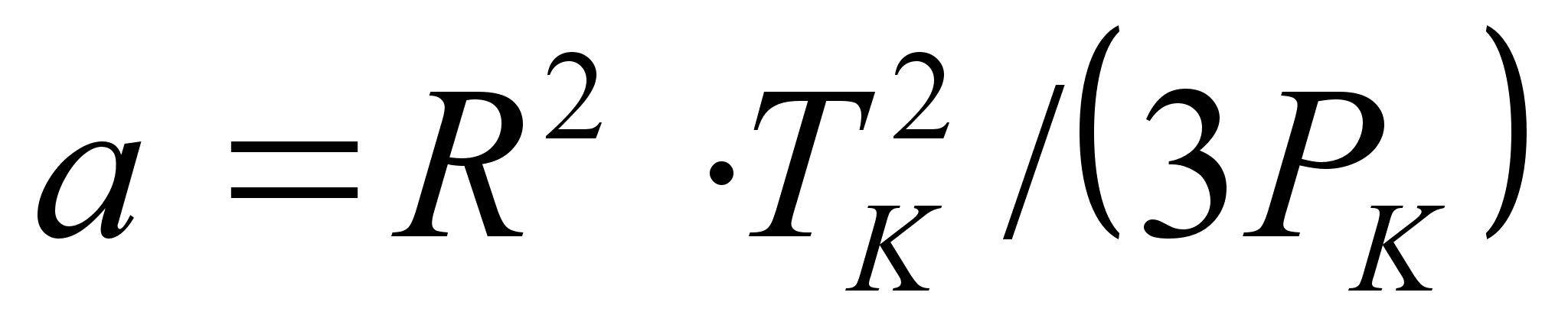 ,
, 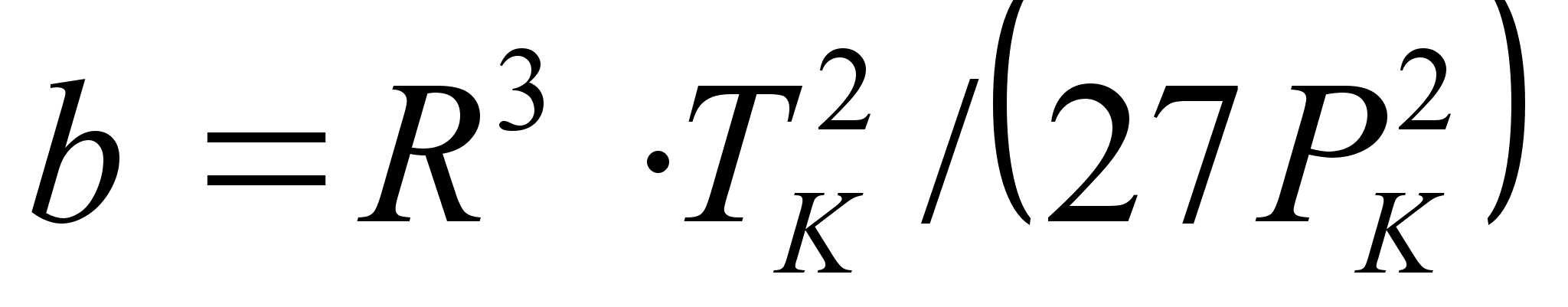 .
.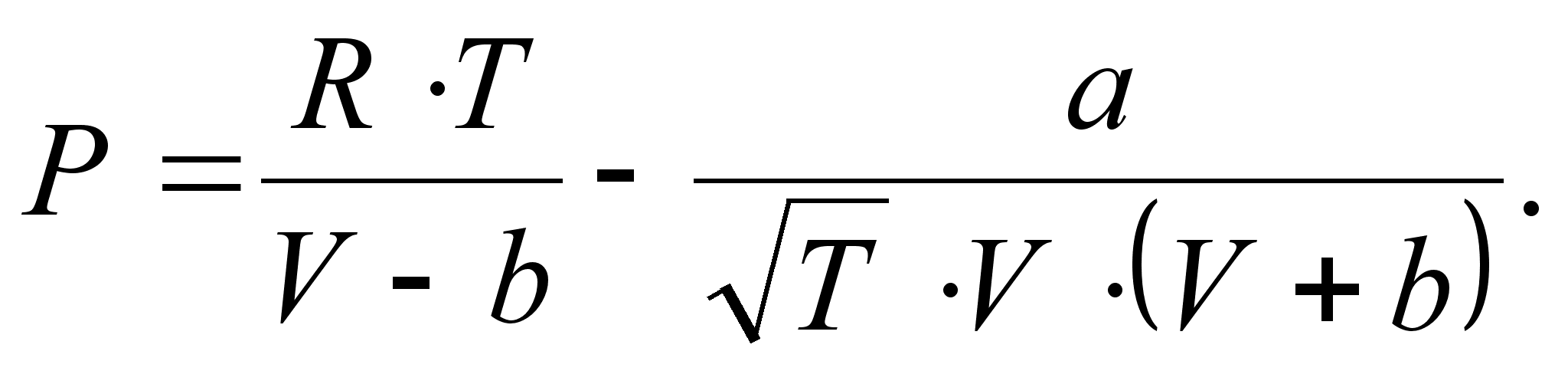
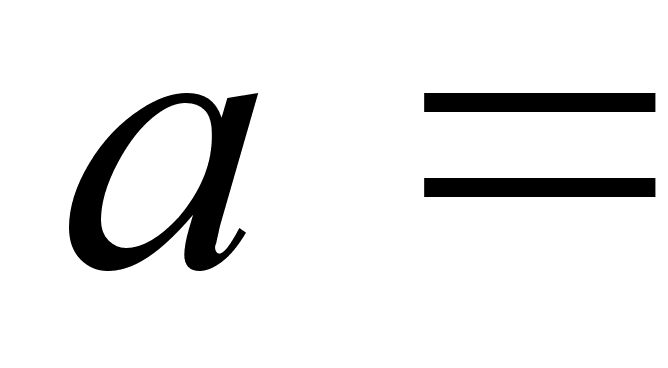 0,42748·R 2 ·T 2,5 k/Pk, b = 0,08664·R·Tk/Pk. Уравнение Редлиха-Квонга считается наилучшим двухконстантным уравнением. При его выводе авторы не руководствовались какими-то определенными теоретическими обоснованиями [8]. Это уравнение следует рассматривать как произвольную, но удачную эмпирическую модификацию предшествующих уравнений состояния.
0,42748·R 2 ·T 2,5 k/Pk, b = 0,08664·R·Tk/Pk. Уравнение Редлиха-Квонга считается наилучшим двухконстантным уравнением. При его выводе авторы не руководствовались какими-то определенными теоретическими обоснованиями [8]. Это уравнение следует рассматривать как произвольную, но удачную эмпирическую модификацию предшествующих уравнений состояния.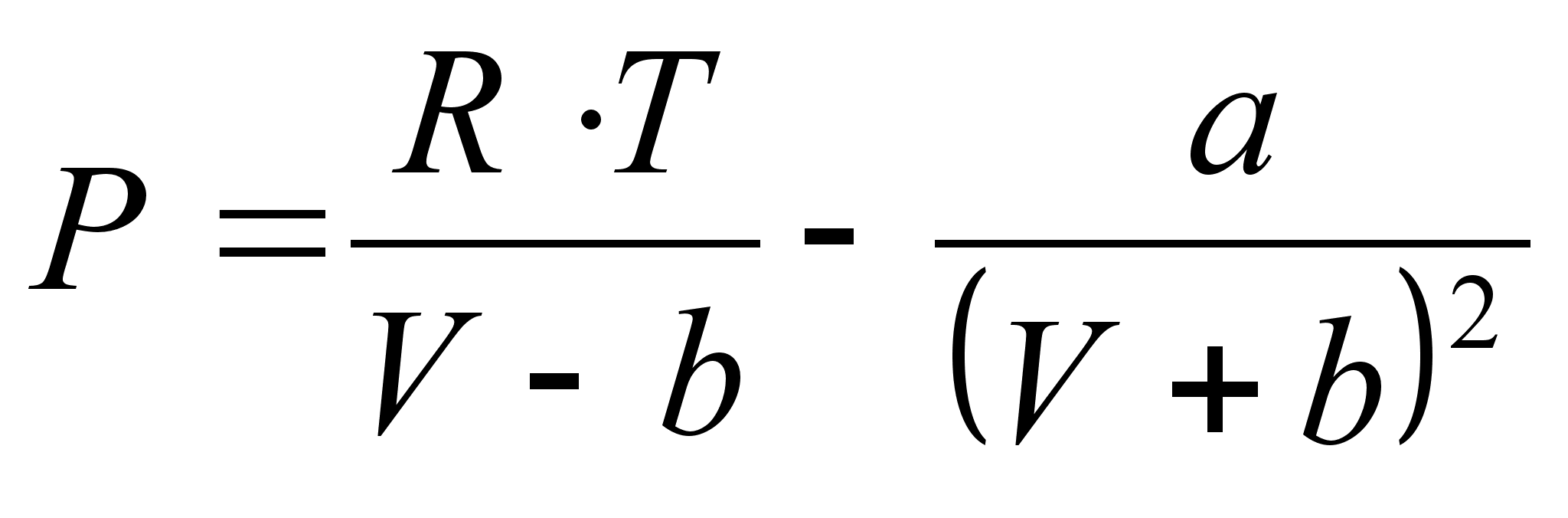 ,
,