Зависимость константы равновесия от давления уравнение планка
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Конспект лекций для студентов биофака ЮФУ (РГУ)
1.7 ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Как было показано выше, протекание самопроизвольного процесса в термодинамической системе сопровождается уменьшением свободной энергии системы (dG 2 Y > 0. Таким образом, условием термодинамического равновесия в закрытой системе является минимальное значение соответствующего термодинамического потенциала :
Изобарно-изотермические (P = const, T = const):
Изохорно-изотермические (V = const, T = const):
Состояние системы с минимальной свободной энергией есть состояние термодинамического равновесия:
Термодинамическим равновесием называется такое термодинамическое состояние системы, которое при постоянстве внешних условий не изменяется во времени, причем эта неизменяемость не обусловлена каким-либо внешним процессом.
Учение о равновесных состояниях – один из разделов термодинамики. Далее мы будем рассматривать частный случай термодинамического равновесного состояния – химическое равновесие. Как известно, многие химические реакции являются обратимыми, т.е. могут одновременно протекать в обоих направлениях – прямом и обратном. Если проводить обратимую реакцию в закрытой системе, то через некоторое время система придет в состояние химического равновесия – концентрации всех реагирующих веществ перестанут изменяться во времени. Необходимо отметить, что достижение системой состояния равновесия не означает прекращения процесса; химическое равновесие является динамическим, т.е. соответствует одновременному протеканию процесса в противоположных направлениях с одинаковой скоростью. Химическое равновесие является подвижным – всякое бесконечно малое внешнее воздействие на равновесную систему вызывает бесконечно малое изменение состояния системы; по прекращении внешнего воздействия система возвращается в исходное состояние. Ещё одним важным свойством химического равновесия является то, что система может самопроизвольно прийти в состояние равновесия с двух противоположных сторон. Иначе говоря, любое состояние, смежное с равновесным, является менее устойчивым, и переход в него из состояния равновесия всегда связан с необходимостью затраты работы извне.
Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации С, парциальные давления P или мольные доли X реагирующих веществ. Для некоторой реакции

соответствующие константы равновесия выражаются следующим образом:
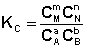 (I.78)
(I.78) 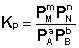 (I.79)
(I.79)
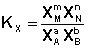 (I.80)
(I.80)
Константа равновесия есть характерная величина для каждой обратимой химической реакции; величина константы равновесия зависит только от природы реагирующих веществ и температуры. Выражение для константы равновесия для элементарной обратимой реакции может быть выведено из кинетических представлений.
Рассмотрим процесс установления равновесия в системе, в которой в начальный момент времени присутствуют только исходные вещества А и В. Скорость прямой реакции V1 в этот момент максимальна, а скорость обратной V2 равна нулю:
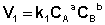 (I.81)
(I.81)
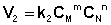 (I.82)
(I.82)
По мере уменьшения концентрации исходных веществ растет концентрация продуктов реакции; соответственно, скорость прямой реакции уменьшается, скорость обратной реакции увеличивается. Очевидно, что через некоторое время скорости прямой и обратной реакции сравняются, после чего концентрации реагирующих веществ перестанут изменяться, т.е. установится химическое равновесие.
Приняв, что V1 = V2, можно записать:
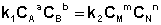 (I.83)
(I.83)
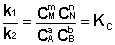 (I.84)
(I.84)
Т.о., константа равновесия есть отношение констант скорости прямой и обратной реакции. Отсюда вытекает физический смысл константы равновесия: она показывает, во сколько раз скорость прямой реакции больше скорости обратной при данной температуре и концентрациях всех реагирующих веществ, равных 1 моль/л.
Теперь рассмотрим (с некоторыми упрощениями) более строгий термодинамический вывод выражения для константы равновесия. Для этого необходимо ввести понятие химический потенциал . Очевидно, что величина свободной энергии системы будет зависеть как от внешних условий (T, P или V), так и от природы и количества веществ, составляющих систему. В случае, если состав системы изменяется во времени (т.е. в системе протекает химическая реакция), необходимо учесть влияние изменения состава на величину свободной энергии системы. Введем в некоторую систему бесконечно малое количество dni молей i-го компонента; это вызовет бесконечно малое изменение термодинамического потенциала системы. Отношение бесконечно малого изменения величины свободной энергии системы к бесконечно малому количеству компонента, внесенному в систему, есть химический потенциал μ i данного компонента в системе:
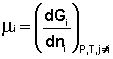 (I.85)
(I.85)
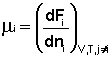 (I.86)
(I.86)
Химический потенциал компонента связан с его парциальным давлением или концентрацией следующими соотношениями:
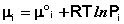 (I.87)
(I.87)
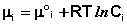 (I.88)
(I.88)
Здесь μ°i – стандартный химический потенциал компонента (Pi = 1 атм., Сi = 1 моль/л.). Очевидно, что изменение свободной энергии системы можно связать с изменением состава системы следующим образом:
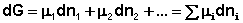 (I.89)
(I.89)
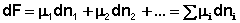 (I.90)
(I.90)
Поскольку условием равновесия является минимум свободной энергии системы (dG = 0, dF = 0), можно записать:
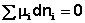 (I.91)
(I.91)
В закрытой системе изменение числа молей одного компонента сопровождается эквивалентным изменением числа молей остальных компонентов; т.е., для приведенной выше химической реакции имеет место соотношение:
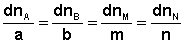 (I.92)
(I.92)
Отсюда можно получить следующее условие химического равновесия в закрытой системе:
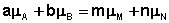 (I.93)
(I.93)
В общем виде условие химического равновесия можно записать следующим образом:
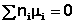 (I.94)
(I.94)
Выражение (I.94) носит название уравнения Гиббса – Дюгема. Подставив в него зависимость химического потенциала от концентрации, получаем:
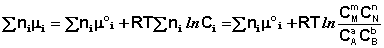 (I.95)
(I.95)
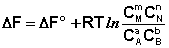 (I.96)
(I.96)
Для изобарно-изотермического процесса аналогичным образом можно получить:
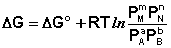 (I.97)
(I.97)
Полученные нами выражения I.96 – I.97 есть изотерма химической реакции . Если система находится в состоянии химического равновесия, то изменение термодинамического потенциала равно нулю; получаем:
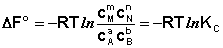 (I.98)
(I.98)
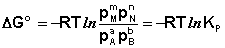 (I.99)
(I.99)
Здесь сi и рi – равновесные концентрации и парциальные давления исходных веществ и продуктов реакции (в отличие от неравновесных Сi и Рi в уравнениях I.96 – I.97).
Поскольку для каждой химической реакции стандартное изменение термодинамического потенциала ΔF° и ΔG° есть строго определенная величина, то произведение равновесных парциальных давлений (концентраций), возведенных в степень, равную стехиометрическому коэффициенту при данном веществе в уравнении химической реакции (стехиометрические коэффициенты при исходных веществах принято считать отрицательными) есть некоторая константа, называемая константой равновесия. Уравнения (I.98, I.99) показывают связь константы равновесия со стандартным изменением свободной энергии в ходе реакции. Уравнение изотермы химической реакции связывает величины реальных концентраций (давлений) реагентов в системе, стандартного изменения термодинамического потенциала в ходе реакции и изменения термодинамического потенциала при переходе из данного состояния системы в равновесное. Знак ΔG (ΔF) определяет возможность самопроизвольного протекания процесса в системе. При этом ΔG° (ΔF°) равно изменению свободной энергии системы при переходе из стандартного состояния (Pi = 1 атм., Сi = 1 моль/л) в равновесное. Уравнение изотермы химической реакции позволяет рассчитать величину ΔG (ΔF) при переходе из любого состояния системы в равновесное, т.е. ответить на вопрос, будет ли химическая реакция протекать самопроизвольно при данных концентрациях Сi (давлениях Рi) реагентов:
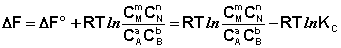 (I.100)
(I.100)
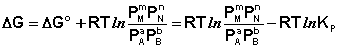 (I.101)
(I.101)
Если изменение термодинамического потенциала меньше нуля, процесс в данных условиях будет протекать самопроизвольно.
1.7.1 Влияние внешних условий на химическое равновесие
При постоянстве внешних условий система может находиться в состоянии равновесия сколь угодно долго. Если изменить эти условия (т.е. оказать на систему какое-либо внешнее воздействие), равновесие нарушается; в системе возникает самопроизвольный процесс, который продолжается до тех пор, пока система опять не достигнет состояния равновесия (уже при новых условиях). Рассмотрим, как влияют на положение равновесия некоторые факторы.
1.7.2 Влияние давления и концентрации
Рассмотрим несколько возможных случаев смещения равновесия.
1. В систему добавлено исходное вещество. В этом случае
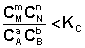 ;
; 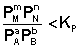 ;
;
По уравнению изотермы химической реакции (I.100 – I.101) получаем: ΔF 0; ΔG > 0. Химическое равновесие будет смещено влево (в сторону расходования продуктов реакции и образования исходных веществ).
3. Изменено общее давление (для реакций в газовой фазе).
Парциальные давления всех компонентов Рi в этом случае изменяются в одинаковой степени; направление смещения равновесия будет определяться суммой стехиометрических коэффициентов Δn.
Учитывая, что парциальное давление газа в смеси равно общему давлению, умноженному на мольную долю компонента в смеси (Рi = РХi), изотерму реакции можно переписать в следующем виде (здесь Δn = Σ(ni) прод – Σ(ni) исх):
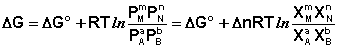 (I.102)
(I.102)
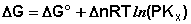 (I.103)
(I.103)
Примем, что Р2 > Р1. В этом случае, если Δn > 0 (реакция идет с увеличением числа молей газообразных веществ), то ΔG > 0; равновесие смещается влево. Если реакция идет с уменьшением числа молей газообразных веществ (Δn изобару Вант-Гоффа :
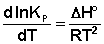 (I.06)
(I.06)
Рассуждая аналогичным образом, для процесса, проходящего в изохорных условиях, можно получить изохору Вант-Гоффа :
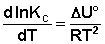 (I.107)
(I.107)
Изобара и изохора Вант-Гоффа связывают изменение константы химического равновесия с тепловым эффектом реакции в изобарных и изохорных условиях соответственно. Очевидно, что чем больше по абсолютной величине тепловой эффект химической реакции, тем сильнее влияет температура на величину константы равновесия. Если реакция не сопровождается тепловым эффектом, то константа равновесия не зависит от температуры.
Экзотермические реакции: ΔH° 0 (ΔU° > 0). В этом случае температурный коэффициент логарифма константы равновесия положителен; повышение температуры увеличивает величину константы равновесия (смещает равновесие вправо).
Графики зависимостей константы равновесия от температуры для экзотермических и эндотермических реакций приведены на рис. I.4.
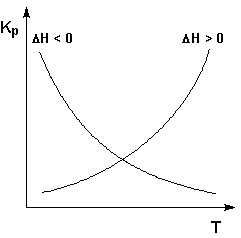
Рис. 1.4 Зависимость константы равновесия от температуры.
Действие рассмотренных нами факторов (давления, концентрации и температуры), равно как и любых других, на систему, находящуюся в состоянии равновесия, обобщает принцип смещения равновесия , называемый также принципом Ле Шателье – Брауна :
Если на систему, находящуюся в состоянии истинного равновесия, оказывается внешнее воздействие, то в системе возникает самопроизвольный процесс, компенсирующий данное воздействие.
Принцип Ле Шателье – Брауна является одним из следствий второго начала термодинамики и применим к любым макроскопическим системам, находящимся в состоянии истинного равновесия.
Copyright © С. И. Левченков, 1996 — 2005.
Зависимость константы равновесия от давления уравнение планка
Большинство химических реакций протекают одновременно в двух направлениях: в сторону образования продуктов реакции (прямая реакция) и в сторону разложения последних (обратная реакция). Вследствие химической обратимости реакции не доходят до конца. Скорость прямой реакции уменьшается, а скорость обратной, напротив, возрастает. Когда эти скорости выравниваются наступает состояние химического равновесия.
Так как химически обратимые реакции до перехода в равновесное состояние протекают с конечными скоростями, то с точки зрения термодинамики они не обратимы. Однако можно мысленно представить, что эти реакции идут бесконечно медленно через смежные равновесные состояния. Тогда к ним можно применить общие условия термодинамического равновесия.
Для гомогенных обратимых реакций экспериментально Гульбергом и Ваге был установлен закон действующих масс. При постоянной температуре отношение произведения равновесных концентраций (или парциальных давлений) продуктов реакции к произведению равновесных концентраций (или парциальных равновесий) исходных веществ есть величина постоянная.
Этот экспериментально установленный закон может быть получен методом термодинамических потенциалов. Рассмотрим реакцию в газовой фазе:
аА(г) + b В ↔ сС + dD
Когда система достигает термодинамического равновесия, то термодинамический потенциал при фиксированных естественных переменных достигает минимума. Равновесие, таким образом, можно охарактеризовать выражением химических потенциалов, когда потенциалы продуктов реакции сравняются с потенциалами исходных веществ:
с μ ( с ) + d μ (D) – a μ (a) — b μ (b) = 0 (6 – 1)
Если естественными переменными являются p и T , то  =
=  , а
, а  = V
= V
Отсюда для систем, подчиняющихся закону идеальных газов, можно получить выражения для μ i
μ i = μ i ° + RTlnCi (6 – 2) 
где μ i ° — стандартный химический потенциал.
Подставляется (6 – 2) в (6 – 1) и перенося постоянные величины в левую часть, получаем
сμ C ° + d μ D ° — a μ A ° — b μ B ° = — RTln 
 (6 – 3)
(6 – 3)
Поскольку в левой части выражение не зависит от концентраций, то выражение под логарифмом является постоянной величиной при постоянной температуре:

Для идеального газа парциальные давления пропорциональны концентрациям, поэтому константа равновесия может быть всегда выражена через равновесные парциальные давления:

Аналогично может быть записано выражение через мольные доли:

Для идеальных газов эти константы связаны между собой соотношением:

где 
Следует обратить внимание, что в полученных соотношениях только KN зависит от общего давления. Она позволяет нам оценивать сдвиг равновесия в газовых реакциях при изменении общего давления. Следует иметь в виду, что давление в этих выражениях складывается из парциальных давлений компонентов системы и не учитывает влияние инертных газов, если они присутствуют в реакционной смеси. Естественно инертный газ «разбавляет» компоненты реакционной смеси и поэтому влияет на KN .
Из уравнения (6 – 3) вытекает связь константы равновесия с ∆ rG °:
 (6 – 4)
(6 – 4)
Это уравнение было впервые получено Вант – Гофором методом циклов и получило название уравнения изотермической химической реакции. Очевидно, в этом уравнении ∆ rGT ° относится к этой температуре, при которой определена Кр. Уравнение изотермической химической реакции позволяет определить константу равновесия при заданных условиях не прибегая к исследованию равновесия. Величина ∆ rGT ° может быть рассчитана на основе термических констант для индивидуальных веществ.
Если заданы концентрации (парциальные давления) отличные от равновесных, то можно записать более общий вид уравнения изотермической химической реакции:

Это выражение позволяет определить направление самопроизвольного процесса.
Уравнение изотермы химической реакции позволяет получить выражение для температурной зависимости константы равновесия.
Запишем уравнение Гиббса – Гельмгольца:

Подставим выражение для  из (6 – 4)
из (6 – 4)
 (6 — 5)
(6 — 5)
Дифференцируем уравнение (6 – 5)

 (6 — 5´)
(6 — 5´)
Из уравнения (6 — 5´) получаем уравнение изобары химической реакции:
 (6 – 6)
(6 – 6)
Если проинтегрировать уравнение (6 – 6) в предположении, что ∆ rHT ° не зависит от температуры, то получим уравнение:

где С – константа интегрирования.
Уравнение (6 – 7) хорошо выполняется в узких интервалах температур и позволяет определить ∆ rGT °.
Для широких интервалов температур ln K р представляют в виде степенных рядов или других аналитических формах:

Такие выражения позволяют рассчитать все термодинамические функции для процессов, для которых данные зависимости получены.
Выражения для термодинамических потенциалов, полученные для идеального газа. Для реальных газов, а особенно для газовых растворов возникают затруднения. Это связано с тем, что расчет концентраций и давлений должен быть проведен исходя из уравнения состояния. Однако для реальных систем единое достаточно простое уравнение состояния получить не удалось.
В связи с этим в термодинамике реальных систем применяется эмпирический метод, предложенный Льюисом. Льюис предложил в уравнениях термодинамики, полученных для идеальных систем заменить давления p на величину летучести f , а концентрации С на активности a .
При такой замене выражения для констант равновесия не меняются по форме. Но этот прием позволяет связать экспериментально найденные свойства реального газа с термодинамическими параметрами.
Летучести и активности – это экспериментальные величины, которые находятся из условия, что для раствора при бесконечном разбавлении или газа при давлении стремящимся к 0 активность приближается к аналитической концентрации, а летучесть к реальному давлению идеального газа. Исходя из этой посылки рассчитываются активности и летучести.
При 1273 К и общем равновесии 30 атм. В равновесной системе
 содержится 17% (по общему)
содержится 17% (по общему)  . Сколько процентов будет содержаться в газе при общем давлении 20 атм.? При каком давлении в газе будет содержаться 25% ? (Газ считать идеальным).
. Сколько процентов будет содержаться в газе при общем давлении 20 атм.? При каком давлении в газе будет содержаться 25% ? (Газ считать идеальным).
В соответствии с законом Авогадро, объёмный процент равен мольному проценту. Следовательно,  при 30 атм. будет равен:
при 30 атм. будет равен:

Отсюда находим 

В отличие от ,  для идеальных газов не зависит от давления. На основании этого находим при 20 атм.
для идеальных газов не зависит от давления. На основании этого находим при 20 атм.

 = 0,125 или 12,5%
= 0,125 или 12,5%
Для 25% 

Следовательно, 
При 2000°С и общем давлении 1 атм. 2% воды диссоцииовано на водород и кислород. Рассчитайте константу равновесия реакции  при этих условиях.
при этих условиях.
ВЛИЯНИЕ ДАВЛЕНИЯ НА КОНСТАНТУ ХИМИЧЕСКОГО
РАВНОВЕСИЯ (УРАВНЕНИЕ ПЛАНКА)
Для установления влияние давления на реакции, протекающие с участием газообразных веществ, воспользуемся константой равновесия, выраженной через равновесные мольные доли, Kх= f(T, p).
Выражение (9), записанное в виде 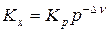 , логарифмируют и полученное выражение с учетом того, что Kр не является функцией давления, дифференцируют по давлению при постоянной температуре
, логарифмируют и полученное выражение с учетом того, что Kр не является функцией давления, дифференцируют по давлению при постоянной температуре
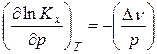 . (18)
. (18)
Считая газы, участвующие в реакции, идеальными, можно из уравнения Менделеева-Клапейрона выразить изменение числа моль газообразных веществ в реакции 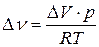 и подставить в уравнение (18). Тогда
и подставить в уравнение (18). Тогда
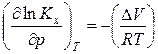 . (19)
. (19)
Уравнения (18) и (19) описывают влияние давления на химическое равновесие в идеальной газовой реакции и называют уравнением Планка. Проведем анализ данного уравнения:
1) если реакция протекает с увеличением объема (количества вещества), то при повышении давления 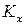 уменьшается. Это означает, что для реакций типа
уменьшается. Это означает, что для реакций типа
А + B = 3C с ростом давления равновесие смещается в сторону исходных веществ;
2) если реакция протекает с уменьшением объема (количества вещества), то при повышении давления увеличивается. Для реакций типа А + 2B = C с ростом давления равновесие смещается в сторону продуктов реакции;
3) если реакция протекает без изменения объема (количества вещества), то при повышении давления не изменяется. Это означает, что для реакций типа
А + B = 2C с ростом давления равновесие не изменяется.
Влияние давления на химическое равновесие в растворе незначительно, так как объем раствора практически не изменяется.
3. растворы и гетерогенные равновесия
3.1. Основные понятия и определения
Термодинамическую систему однородную по физическому строению и химическим свойствам во всех точках, называют гомогенной.
Термодинамическую систему, состоящую из различных по физическим или химическим свойствам частей, отделенных друг от друга поверхностями раздела, называют гетерогенной.
Любая гетерогенная система состоит из нескольких фаз. Фаза – это гомогенная часть гетерогенной системы, ограниченная поверхностью раздела, при переходе через которую свойства системы меняются скачкообразно. Системы делятся на одно–, двух–, трехфазные и т.д.
Каждая система состоит из одного или нескольких веществ, называемых компонентами. Вещества, образующие термодинамическую систему, могут находиться в различных агрегатных состояниях: газообразном, жидком, твердом. Числом независимых компонентов называют наименьшее число индивидуальных компонентов, необходимое для образования данной системы, которое равно общему числу индивидуальных веществ, входящих в данную систему, за вычетом числа уравнений, связывающих равновесные концентрации этих веществ. По числу компонентов различают одно–, двух–, трех– и т.д. компонентные системы.
Любая система характеризуется внешними ( 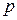 ,
, 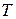 , и т.д.) и внутренними (концентрационными) параметрами состояния или концентрациями, которые определяют равновесный состав фаз.
, и т.д.) и внутренними (концентрационными) параметрами состояния или концентрациями, которые определяют равновесный состав фаз.
Число концентрационных параметров данной фазы равно числу независимых компонентов, входящих в ее состав фазы, за вычетом единицы (так как, например, если система состоит их двух компонентов, то концентрацию второго компонента можно определить, зная концентрацию первого).
Число независимых термодинамических параметров состояния данной системы, произвольное изменение которых в определенных пределах не вызывает исчезновения одних и образование других фаз называют числом термодинамических степеней свободы или вариантностью системы. По числу термодинамических степеней свободы системы разделяются на инвариантные ( 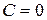 ), моновариантные (
), моновариантные ( 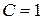 ), дивариантные (
), дивариантные ( 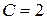 ) и т.д.
) и т.д.
3.2. Термодинамика растворов
3.2.1. ОСНОВНЫЕ ПОНЯТИЯ
Раствором называют гомогенную однофазную систему, состоящую минимум из двух независимых компонентов, в каждом элементарном объеме которого одинаковые физические, химические и термодинамические свойства. В жидких растворах обычно различают растворитель – это вещество, которое имеется в растворе в избытке, и растворенные вещества, хотя все компоненты раствора термодинамически равноценны. Компоненты, находящиеся в растворе в меньшем количестве называют растворенными веществами.
Растворы подразделяют на идеальные и реальные. Идеальным называют раствор, все компоненты которого характеризуются одинаковой формой и размером молекул и одинаковой энергией межмолекулярных взаимодействий. Идеальные растворы встречаются довольно редко. Это гомогенные смеси близких по физико-химическим свойствам веществ (смеси оптических изомеров, соседних членов одного и того же гомологического ряда). Моделью идеального газового раствора является смесь идеальных газов.
Большинство растворов являются реальными, их компоненты отличаются либо по форме, либо по размерам, либо по энергии межмолекулярных взаимодействий.
Все свойства растворов подразделяют на экстенсивные и интенсивные.
Экстенсивные свойства, зависят как от общей массы раствора, так и от его состава, например 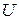 ,
, 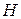 ,
, 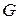 ,
, 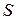 ,
, 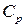 . Эти свойства относятся ко всему раствору, как единому целому, а не к отдельным его компонентам.
. Эти свойства относятся ко всему раствору, как единому целому, а не к отдельным его компонентам.
Интенсивные свойства, зависят только от состава раствора и не зависят от его общей массы, например, давление насыщенного пара.
Для характеристики растворов используют средние мольные и парциальные мольные свойства.
Среднее мольное свойство – экстенсивное свойство 1 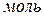 раствора.
раствора.
Парциальное мольное свойство 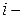 го компонента – это частная производная от экстенсивного свойства раствора по числу моль этого компонента (
го компонента – это частная производная от экстенсивного свойства раствора по числу моль этого компонента ( 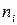 ) при постоянном числе моль всех остальных компонентов и внешних параметрах ( , ).
) при постоянном числе моль всех остальных компонентов и внешних параметрах ( , ).
Среди парциальных молярных величин наибольшее значение имеет парциальная мольная энергия Гиббса, которая называется химическим потенциалом 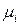 .
.
Химический потенциал является интенсивным свойством раствора.
3.2.2. УСЛОВИЕ РАВНОВЕСИЯ В ГОМОГЕННЫХ РАСТВОРАХ
Равновесие в гомогенном идеальном растворе выражает уравнение Гиббса–Дюгема:

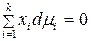 , (1)
, (1)
которое для двухкомпонентного раствора записывается в виде
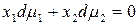 .
.
Равновесие в гомогенном реальном растворе выражает уравнение Дюгема–Маргулеса:

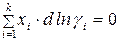 . (2)
. (2)
Для двухкомпонентного раствора условие равновесия записывается:
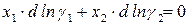 .
.
3.3. гетерогенные равновесия
3.3.1. равновесие в гетерогенной системе. Правило фаз гиббса
Условием термодинамического равновесия в гетерогенной системе является равенство химических потенциалов каждого компонента во всех фазах при 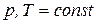 или
или 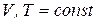 :
:
 Для
Для 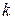 –компонентной и
–компонентной и 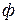 –фазной системы при условие термодинамического равновесия (теорема Гиббса) выражается системой уравнений
–фазной системы при условие термодинамического равновесия (теорема Гиббса) выражается системой уравнений
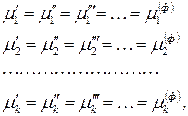 (3)
(3)
где индекс 1, 2,…, – номер компонента;
¢,²,¢¢¢,…, – номер фазы.
 В равновесной системе связь между числом фаз, числом компонентов и числом термодинамических степеней свободы выражает основной закон фазового равновесия или правило фаз Гиббса:
В равновесной системе связь между числом фаз, числом компонентов и числом термодинамических степеней свободы выражает основной закон фазового равновесия или правило фаз Гиббса:
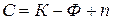 , (4)
, (4)
где 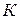 – число независимых компонентов;
– число независимых компонентов;
n – число внешних параметров, влияющих на состояние равновесия.
Если на систему влияют два внешних параметра ( и ), то правило фаз Гиббса записывается
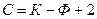 .
.
Применим правило фаз Гиббса к анализу диаграммы состояния однокомпонентной системы, например, воды. В области средних давлений и средних температур вода может находиться в жидком, твердом (лед) и газообразном (пар) состояниях.
 На рис. 1:
На рис. 1:
плоскость ОВС отвечает состоянию воды;
АОС – состоянию пара;
АОВ – состоянию льда;
О – тройная точка;
ОВ – кривая плавления;
ОС – кривая испарения;
ОА – кривая возгонки;
ОD – кривая давления насыщенного пара над переохлажденной водой.
Определим число степеней свободы в точках 1, 2 и тройной точке О.
В точке 1 вода находится в жидком состоянии, следовательно, число фаз 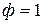 , тогда
, тогда
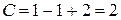 .
.
Число степеней свободы равно двум или система дивариантна. Это означает, что можно произвольно в определенных пределах изменять два параметра: давление и температуру, при этом число и вид фаз системы не изменится.
Точка 2 находится на кривой испарения, следовательно, в равновесии находятся две фазы: жидкость и пар. Тогда
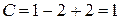 .
.
Система моновариантна, следовательно, возможно изменение одного параметра: температуры или давления, при котором число и вид фаз системы не изменится.
В тройной точке О в равновесии находятся три фазы: вода, лед и пар, тогда
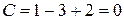 .
.
Система нонвариантна. Это означает, что три фазы могут находиться в равновесии только при определенных условиях.
3.3.2. Уравнение состояния однокомпонентной двухфазной системы
Состояние однокомпонентной двухфазной системы характеризуется уравнением Клаузиуса–Клапейрона:
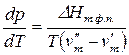
где 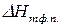 – мольная теплота фазового перехода;
– мольная теплота фазового перехода;
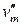 ,
, 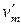 – мольный объем вещества в фазе ² и ¢ соответственно.
– мольный объем вещества в фазе ² и ¢ соответственно.
Для процесса возгонки и испарения можно допустить, что 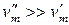 . При условии, что паровую фазу можно считать идеальной, в соответствии с уравнением Менделеева–Клапейрона
. При условии, что паровую фазу можно считать идеальной, в соответствии с уравнением Менделеева–Клапейрона 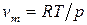 , тогда уравнение Клаузиуса–Клапейрона запишется:
, тогда уравнение Клаузиуса–Клапейрона запишется:

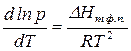 . (5)
. (5)
Разделив переменные и проинтегрировав в определенных пределах в узком интервале температур, считая постоянной величиной, получим
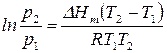 .
.
На основе полученного уравнения можно рассчитать:
1) температуру кипения вещества  под давлением
под давлением  , если известна температура кипения
, если известна температура кипения 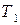 этого вещества под давлением
этого вещества под давлением 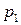 и величина средней мольной теплоты испарения;
и величина средней мольной теплоты испарения;
2) давление насыщенного пара индивидуального вещества при температуре , если известно давление насыщенного пара 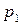 при температуре и средняя мольная теплота испарения;
при температуре и средняя мольная теплота испарения;
3) среднюю мольную теплоту испарения или возгонки вещества, если известны значения давления насыщенного пара вещества при двух температурах.
3.3.3. фазовое равновесие жидкость-пар
Пусть существуют в состоянии равновесия жидкость и выделяющийся из нее пар. Если жидкий раствор является идеальным, то во всем интервале концентраций растворитель и растворенное вещество подчиняются закону Рауля:

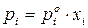 , (6)
, (6)
где 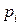 – давление насыщенного пара го компонента над раствором;
– давление насыщенного пара го компонента над раствором;
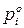 – давление насыщенного пара над индивидуальным м компонентом при температуре раствора.
– давление насыщенного пара над индивидуальным м компонентом при температуре раствора.
Для двухкомпонентного раствора можно записать:
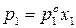 ;
; 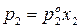 .
.
Так как 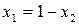 , а общее давление над раствором складывается из парциальных давлений компонентов
, а общее давление над раствором складывается из парциальных давлений компонентов 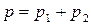 , то
, то
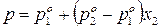 .
.
Таким образом, зависимость давления насыщенного пара компонентов и общего давления пара от состава идеального раствора является линейной (диаграмма 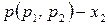 на рис. 2).
на рис. 2).
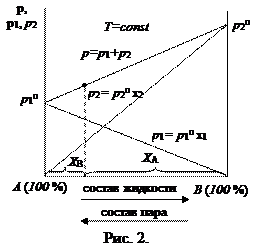
На практике чаще приходится встречаться с неидеальными растворами, которые не подчиняются закону Рауля. Для описания зависимости давления насыщенного пара компонента от состава реального раствора в закон Рауля вводится коэффициент активности:
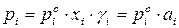 .
.
Для двухкомпонентного раствора общее давление смеси равно:
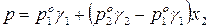 .
.
Поскольку коэффициенты активности 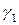 и
и 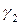 зависят от состава раствора, то зависимость от
зависят от состава раствора, то зависимость от 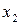 представляет собой кривую. Отклонения давления пара от линейной зависимости в сторону больших значений называют положительными, а в сторону меньших значений – отрицательными отклонениями от закона Рауля (рис. 3). Отклонения
представляет собой кривую. Отклонения давления пара от линейной зависимости в сторону больших значений называют положительными, а в сторону меньших значений – отрицательными отклонениями от закона Рауля (рис. 3). Отклонения
зависят от относительной величины энергии взаимодействия молекул жидкой смеси.

 Иногда отклонения бывают настолько велики, что на кривых давление (температура) – состав появляется ярко выраженный экстремум (минимум или максимум).
Иногда отклонения бывают настолько велики, что на кривых давление (температура) – состав появляется ярко выраженный экстремум (минимум или максимум).
Точки на диаграммах 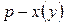 и
и 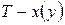 , которым отвечает максимум давления насыщенного пара (минимум температуры кипения) называют положительными (рис. 4а), а если минимум давления (максимум температуры кипения) – отрицательными азеотропами (рис. 4б).
, которым отвечает максимум давления насыщенного пара (минимум температуры кипения) называют положительными (рис. 4а), а если минимум давления (максимум температуры кипения) – отрицательными азеотропами (рис. 4б).
Как видно из рис. 4, в точке азеотропа состав жидкой и паровой фаз одинаков. Бинарные смеси, которые содержат азеотроп, называют азеотропными.
Состав равновесного с жидким раствором пара определяется согласно закону Дальтона:

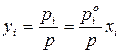 – для идеального раствора;
– для идеального раствора;

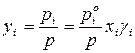 – для реального раствора.
– для реального раствора.
Из анализа последних уравнений видно, что рассчитать равновесный состав пара можно только для идеального раствора заданного состава. Для расчета равновесного состава паровой фазы реального раствора необходимо знать коэффициенты активности, численные значения которых определяют только экспериментальным путем.
Для представления данных по фазовому равновесию жидкость-пар кроме рассмотренных диаграмм применяют также диаграммы 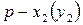 ,
, 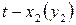 ,
, 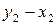 .
.

Для разделения жидких бинарных смесей используют методы перегонки и ректификации (многократной перегонки), которые основаны на различие составов жидкой и паровой фаз. Азеотропные смеси этими методами разделить невозможно. Для разделения азеотропных смесей необходимо создать условия, устраняющие азеотроп. Например, изменить значения внешних параметров ( , ) или добавить к бинарной смеси третий компонент, в присутствии которого азеотроп отсутствует.
Эбуллиоскопия
Эбуллиоскопия – явление повышения температуры кипения раствора нелетучего вещества по сравнению с температурой кипения чистого растворителя.
Основной закон эбуллиоскопии записывается:

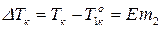 , (7)
, (7)
где 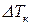 – повышение температуры кипения;
– повышение температуры кипения; 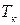 ,
, 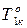 – температура кипения раствора нелетучего вещества и чистого растворителя;
– температура кипения раствора нелетучего вещества и чистого растворителя;
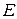 – эбуллиоскопическая константа (для воды = 0,52 (град·кг)/моль)
– эбуллиоскопическая константа (для воды = 0,52 (град·кг)/моль)

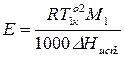 , (8)
, (8)
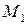 – молекулярная масса растворителя;
– молекулярная масса растворителя; 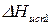 – теплота испарения растворителя;
– теплота испарения растворителя; 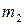 – моляльная концентрация растворенного вещества.
– моляльная концентрация растворенного вещества.
3.3.4. Фазовое равновесие твердое тело-жидкость. Уравнение шредера
Предположим, что в состоянии термодинамического равновесия существуют раствор твердого вещества в жидком растворителе и кристаллы данного твердого вещества. Равновесие в такой системе описывается уравнением Шредера:
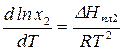 ,
,
где – мольная доля растворенного вещества;
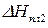 – теплота плавления твердого вещества.
– теплота плавления твердого вещества.
После интегрирования получаем:
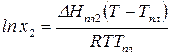 ,
,
где 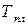 – температура плавления чистого твердого вещества.
– температура плавления чистого твердого вещества.
Полученное уравнение справедливо для растворов, близких по свойствам к идеальным, поэтому уравнение Шредера позволяет рассчитывать растворимость только малорастворимых веществ.
Криоскопия
Криоскопия – явление понижения температуры замерзания раствора нелетучего вещества по сравнению с температурой замерзания чистого растворителя.
Основной закон криоскопии записывается:
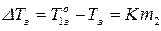 , (9)
, (9)
где 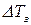 – понижение температуры замерзания;
– понижение температуры замерзания;
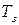 – температура замерзания раствора нелетучего вещества;
– температура замерзания раствора нелетучего вещества;
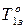 – температура замерзания чистого растворителя;
– температура замерзания чистого растворителя;
– криоскопическая константа (для воды = 1,86 (град·кг)/моль)

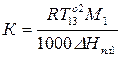 , (10)
, (10)
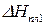 – теплота плавления растворителя.
– теплота плавления растворителя.
Методы криоскопии и эбуллиоскопии часто применяют для расчета молекулярной массы растворенного вещества.
3.3.5. фазовое равновесие жидкость-жидкость
Пусть в сосуде находятся две жидкости, практически не смешивающиеся друг с другом, например, вода и хлороформ. Если ввести в эту систему небольшое количество третьего вещества, например йода, то оно вполне определенным образом распределится между двумя жидкими фазами. В соответствии с законом распределения Нернста: при установлении равновесия отношение концентраций распределяющегося вещества в двух несмешивающихся жидкостях есть величина постоянная при постоянной температуре:

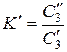 , (11)
, (11)
где 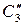 ,
, 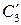 – равновесные концентрации распределяющегося вещества в двух соприкасающихся фазах;
– равновесные концентрации распределяющегося вещества в двух соприкасающихся фазах;
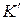 – коэффициент распроеделения.
– коэффициент распроеделения.
 Закон распределения Нернста лежит в основе процесса жидкостной экстракции – извлечения вещества из раствора при помощи другого растворителя, практически не смешивающегося с первым. Экстракция широко применяется в промышленности.
Закон распределения Нернста лежит в основе процесса жидкостной экстракции – извлечения вещества из раствора при помощи другого растворителя, практически не смешивающегося с первым. Экстракция широко применяется в промышленности.
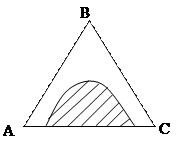
Состояние трехкомпонентных систем с ограниченной растворимостью принято изображать с помощью треугольных диаграмм. На рисунке приведена диаграмма растворимости в трехкомпонентной системе с одной парой ограниченно смешивающихся компонентов 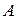 и
и 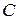 . Вершины треугольника соответствуют содержанию 100 % компонентов ,
. Вершины треугольника соответствуют содержанию 100 % компонентов , 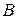 и . Любая точка внутри треугольника выражает состав трехкомпонентной системы. Заштрихованная плоскость соответствует области гетерогенного (двухфазного) состояния системы. Линия, ограничивающая гетерогенную область, называется изотермой взаимной растворимости.
и . Любая точка внутри треугольника выражает состав трехкомпонентной системы. Заштрихованная плоскость соответствует области гетерогенного (двухфазного) состояния системы. Линия, ограничивающая гетерогенную область, называется изотермой взаимной растворимости.
4. химическая кинетика
Химическая кинетика – наука о скорости протекания химической реакции.
Скорость химической реакции – изменение концентрации одного из реагирующих веществ в единицу времени.
Поскольку в реакциях вещества участвуют в стехиометрических соотношениях, за скорость реакции может быть принята производная от концентрации любого из реагирующих веществ по времени:

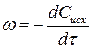 , (1)
, (1)
где 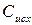 – концентрация исходных веществ;
– концентрация исходных веществ;
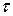 – время.
– время.
Концентрация исходных веществ убывает, поэтому перед производной стоит знак минус.

 Если в качестве одного из реагирующих веществ выбран продукт реакции, то
Если в качестве одного из реагирующих веществ выбран продукт реакции, то
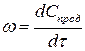 . (2)
. (2)
Скорость реакции в момент времени равна тангенсу угла наклона касательной, проведенной к кривой зависимости 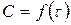 в точке, соответствующей времени :
в точке, соответствующей времени :
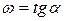 .
.
4.1. основной закон химической кинетики.
http://www.trotted.narod.ru/physchem/lec-6.htm
http://helpiks.org/8-97511.html