Зависимость скорости реакции от температуры определяется уравнением аррениуса
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Конспект лекций для студентов биофака ЮФУ (РГУ)
2.1 СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
2.1.9 Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Я. Г. Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ . Математически правило Вант-Гоффа можно записать следующим образом:
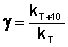 (II.29)
(II.29)
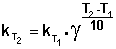 (II.30)
(II.30)
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
2.1.10 Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. С. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом:
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е’А выше, нежели энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
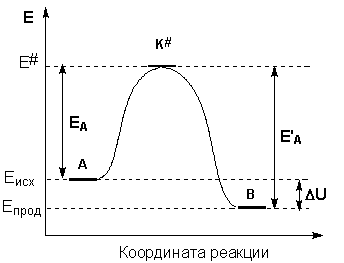
Рис. 2.5 Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.6):
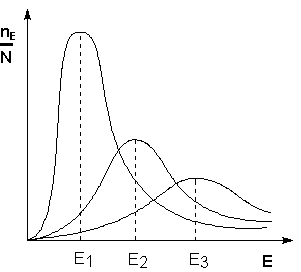
Рис. 2.6 Распределение частиц по энергии
Здесь nЕ/N – доля частиц, обладающих энергией E;
Ei — средняя энергия частиц при температуре Ti (T1 уравнения Аррениуса . Согласно уравнению изобары Вант-Гоффа,
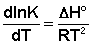 (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
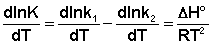 (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
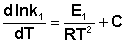 (II.33)
(II.33)
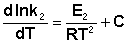 (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации :
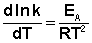 (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
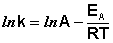 (II.36)
(II.36)
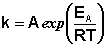 (II.37)
(II.37)
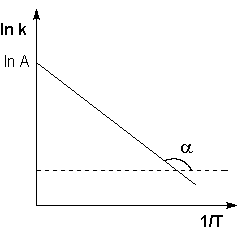
Рис. 2.7 Зависимость логарифма константы скорости химической
реакции от обратной температуры.
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
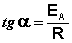 (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
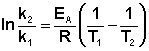 (II.39)
(II.39)
Copyright © С. И. Левченков, 1996 — 2005.
Скорость химической реакции
Говорить об осуществимости процесса можно по изменению энергии Гибсса системы. Но данная величина не отражает настоящую возможность протекания, механизм и скорость химической реакции.
Понятие скорости химической реакции
Для полноценного представления химической реакции, надо иметь знания о том, какие существуют временные закономерности при ее осуществлении, т.е. скорость химической реакции и ее детальный механизм.
Определение скорости химической реакции:
Скорость химической реакции — это изменение концентрации реагирующих веществ в единицу времени
Скорость и механизм реакции изучает химическая кинетика – наука о химическом процессе.
С точки зрения химической кинетики, реакции можно классифицировать на простые и сложные.
Простые реакции – процессы, протекающие без образования промежуточных соединений. По количеству частиц, принимающих в ней участие, они делятся на мономолекулярные, бимолекулярные, тримолекулярные. Соударение большего чем 3 числа частиц маловероятно, поэтому тримолекулярные реакции достаточно редки, а четырехмолекулярные — неизвестны.
Сложные реакции – процессы, состоящие из нескольких элементарных реакций.
Любой процесс протекает с присущей ему скоростью, которую можно определить по изменениям, происходящим за некий отрезок времени.
Среднюю скорость химической реакции выражают изменением количества вещества n израсходованного или полученного вещества в единице объема V за единицу времени t.
Если вещество расходуется, то ставим знак «-», если накапливается – «+»
При постоянном объеме:
Единица измерения скорости реакции — моль/л·с
В целом, υ — величина постоянная и не зависит от того, за каким участвующим в реакции веществом, мы следим.
Зависимость концентрации реагента или продукта от времени протекания реакции представляют в виде кинетической кривой, которая имеет вид:

Вычислять υ из экспериментальных данных удобнее, если указанные выше выражения преобразовать в следующее выражение:
| υ = — ΔC/Δt [моль/л·с] |  |
Закон действующих масс. Порядок и константа скорости реакции
Одна из формулировок закона действующих масс звучит следующим образом:
Скорость элементарной гомогенной химической реакции прямо пропорциональна произведению концентраций реагентов.
Т.е. скорость прямой химической реакции зависит от концентраций исходных веществ.
Если исследуемый процесс представить в виде:
а А + b В = продукты
то скорость химической реакции можно выразить кинетическим уравнением:
а и b – стехиометрические коэффициенты простой реакции,
k – константа скорости реакции.
Химический смысл величины константы скорости реакции k — это скорость реакции при единичных концентрациях.
То есть, если концентрации веществ А и В равны 1, то υ = k.
Надо учитывать, что в сложных химических процессах коэффициенты а и b не совпадают со стехиометрическими.
Закон действующих масс выполняется при соблюдении ряда условий:
- Реакция активируется термично, т.е. энергией теплового движения молекул.
- Концентрация реагентов распределена равномерно.
- Свойства и условия среды в ходе процесса не меняются.
- Свойства среды не должны влиять на k.
К сложным процессам закон действия масс применить нельзя!
Это можно объяснить тем, что сложный процесс состоит из нескольких элементарных стадий, и его скорость будет определяться не суммарной скоростью всех стадий, лишь одной самой медленной стадией, которая называется лимитирующей.
Каждая реакция имеет свой порядок. Определяют частный (парциальный) порядок по реагенту и общий (полный) порядок.
Например, в выражении скорости химической реакции для процесса
а А + b В = продукты
a – порядок по реагенту А
Для простых процессов порядок реакции указывает на количество реагирующих частиц (совпадает со стехиометрическими коэффициентами) и принимает целочисленные значения.
Для сложных процессов порядок реакции не совпадает со стехиометрическими коэффициентами и может быть любым.
Факторы, влияющие на скорость химической реакции
Определим факторы, влияющие на скорость химической реакции υ:
1. Зависимость скорости реакции от концентрации реагирующих веществ
определяется законом действующих масс:
Очевидно, что с увеличением концентраций реагирующих веществ, скорость реакции υ увеличивается, т.к. увеличивается число соударений между участвующими в химическом процессе веществами.
Причем, важно учитывать порядок реакции:
если это n = 1 по некоторому реагенту, то ее скорость прямо пропорциональна концентрации этого вещества.
Если по какому-либо реагенту n = 2, то удвоение его концентрации приведет к росту скорости реакции в 2 2 = 4 раза.
А увеличение концентрации в 3 раза ускорит реакцию в 3 2 = 9 раз.
2. Зависимость скорости реакции от давления
Справедлива для веществ в газообразном состоянии и определяется уравнением Клапейрона – Менделеева , которое связывает концентрацию и давление:
Таким образом, изменение концентрации в системе, а следовательно и скорости реакции имеет прямую зависимость от изменения давления.
Например, для реакции первого порядка, увеличение давления в 2 раза вызовет рост концентрации вещества в 2 раза, что непременно изменит скорость реакции υ – она станет в 2 раза больше.
3. Зависимость скорости реакции от площади поверхности
Для гетерогенных реакций скорость реакции зависит от площади соприкосновения частиц:
где S — площадь соприкосновения частиц, мм 2 ,
Δn — изменение количества веществ, принимающих участие в реакции (исходных веществ или продуктов реакции), моль;
Δt — промежуток времени, с;
Единица измерения скорости гетерогенной реакции, моль/м 2 ⋅с.
Таким образом, вещества реагируют быстрее, если площадь поверхности, на которой может происходить взаимодействие веществ больше.
Растворяя вещество, мы уменьшаем его размеры до размеров молекулы, увеличивая тем самым площадь поверхности.
Поэтому химические процессы между веществами, находящимися в растворенном, жидком или газообразном состоянии имеют большую скорость, чем взаимодействия между твердыми веществами.
4. Зависимость скорости реакции от природы вещества .
В этом случае, большое значение имеет строение электронной оболочки атома, тип химической связи и ее прочность в молекулах, структура вещества, прочность его кристаллической решетки.
Например, натрий будет активнее взаимодействовать с водой, чем олово. Поэтому и скорость взаимодействия натрия с водой выше скорости взаимодействия олова с водой.
5. Зависимость скорости реакции от температуры
определяется правилом Вант-Гоффа и уравнением Аррениуса
Повышая температуру, мы сообщаем молекулам дополнительную энергию (увеличивая, тем самым, энергию активации), которая способствует протеканию реакции.
Поэтому, при повышении температуры скорость химической реакции увеличивается.
Сванте Аррениус в 1889 году, изучая зависимость скорости реакции υ от температуры, установил, что большинство химических процессов подчиняются уравнению:

где k — константа скорости реакции
Еа -энергия активации – минимальная (критическая) энергия, необходимая для осуществления реакции, единица измерения Дж/моль
Т — абсолютная температура
R – газовая постоянная, R = 8,314 Дж/моль·град
A — предэкспоненциальный множитель (частотный фактор), единица измерения совпадает с k. Эта константа выражает вероятность того, что при столкновении молекулы будут ориентированы так, чтобы взаимодействие было возможно.
Если известна константа скорости k при одной температуре Т1, а требуется найти константу скорости k при некой другой температуре Т2, то это легко сделать, если взять логарифм уравнения Аррениуса при Т1 и Т2:
Вычитая второе равенство из первого, получаем:

Уравнение Аррениуса при определении скорости химической реакции (в случае, если υ описывается степенным уравнением) , принимает вид:

Если принять, что концентрации веществ А и В постоянны и прологарифмировать данное выражение, то получим следующее выражение:
Правило Вант-Гоффа
Также удобно пользоваться эмпирическим правилом, которое сформулировал Якоб Вант-Гофф:
увеличение температуры на каждые 10 градусов, приводит к росту скорости реакции в 2 – 4 раза.
Правило Вант-Гоффа имеет математическое выражение:

γ — температурный коэффициент реакции, значения которого лежат в интервале от 2 до 4.
Приведем пример применения правила Вант-Гоффа.
υT1/υT2 = 3 2 = 9. Это означает, что υ возросла в 9 раз.
6. Зависимость скорости реакции от присутствия катализатора
Катализ – это любое изменение скорости реакции под действием катализатора. Он может быть положительным и отрицательным. Суть катализа – генерирование активного субстрата или реагента с участием катализаторов.
Катализатор представляет собой вещество, которое селективно ускоряет химическую реакцию, вступая при этом в промежуточную стадию, но регенирируясь к ее концу (к моменту образования конечных продуктов). Например, в биохимической среде в качестве катализаторов выступают ферменты.
Если такое вещество замедляет химическую реакцию, то оно называется ингибитором.
Влияние катализатора на скорость реакции основывается на том, что он изменяет энергию активации Еа. Понижение энергии активации под действием катализатора схематично представлено на рисунке ниже:
Видно, что веществам А и В требуется большое количество энергии, чтобы образовать конечные продукты. Но в присутствии катализатора для получения конечных продуктов требуется гораздо меньше энергии, т.к. идет понижение полной энергии активации, и тем самым, увеличение скорости реакции.
Обращаю ваше внимание на то, что энергии как начальных, так и конечных веществ остаются одинаковыми в обеих реакциях.
ВАНТ-ГОФФА ПРАВИЛО
ВАНТ-ГОФФА ПРАВИЛО. Почти все химические реакции при повышении температуры идут быстрее. Зависимость скорости реакции от температуры описывается уравнением Аррениуса:
k = Ae –E a/RT, где k – константа скорости реакции, А – не зависящая от температуры константа (ее называют предэкспоненциальным множителем), Еа– энергия активации, R – газовая постоянная, Т – абсолютная температура. В школьных учебниках зависимость скорости реакции от температуры определяют в соответствии с так называемым «правилом Вант-Гоффа», которое в 19 в. сформулировал голландский химик Якоб Вант-Гофф. Это чисто эмпирическое правило, т.е. правило, основанное не на теории, а выведенное из опытных данных. В соответствии с этим правилом, повышение температуры на 10° приводит к увеличению скорости в 2–4 раза. Математически эту зависимость можно выразить уравнением v2v1 = g (T2 – T 1 )/10 , где v1 и v2– скорости реакции при температурах Т1 и Т2; величина g называется температурным коэффициентом реакции. Например, если g = 2, то при Т2– Т1 = 50 о v2/v1 = 2 5 = 32, т.е. реакция ускорилась в 32 раза, причем это ускорение никак не зависит от абсолютных величин Т1 и Т2, а только от их разности.
Однако из уравнения Аррениуса следует, что температурный коэффициент реакции зависит как от энергии активации, так и от абсолютной температуры. Для данной реакции с определенным значением Еа ускорение при повышении температуры на 10° будет тем больше, чем ниже температура. Это почти очевидно и без расчетов: повышение температуры от 0 до 10° С должно сказаться на скорости реакции значительно сильнее, чем такое же повышение температуры, например, от 500 до 510° С.
С другой стороны, для данного температурного интервала ускорение реакции будет тем сильнее, чем больше ее энергия активации. Так, если энергия активации реакции мала, то такая реакция идет очень быстро, и при повышении температуры на 10° С ее скорость почти не изменяется. Для таких реакций температурный коэффициент намного меньше 2. Для реакций же с большой энергией активации, которые при невысоких температурах идут медленно, ускорение при повышении температуры на 10° С может значительно превысить 4-кратное.
Например, реакция диоксида углерода со щелочным раствором с образованием гидрокарбонат-иона (СО2 + ОН® НСО3 – ) имеет энергию активации 38,2 кДж/моль, поэтому при повышении температуры, например, от 50 до 60° С эта реакция ускорится всего в 1,5 раза. В то же время реакция распада этилбромида на этилен и бромоводород (С2Н5Вr ® С2Н4 + НВr) с энергией активации 218 кДж/моль ускорится при повышении температуры от 100 до 110 o С в 6,3 раза (правда, в этом интервале температур реакция идет очень медленно). Кинетика реакции атомов водорода с этаном H + C2H6 ® H2 + C2H5 была изучены в широком температурном интервале – от 300 до 1100 К (27–827° С). Для этой реакции Eа = 40,6 кДж/моль. Следовательно, повышение температуры на 10° вызовет увеличение скорости реакции в 1,7 раза в интервале 300–310 K и только в 1,04 раза в интервале 1090–1100 K. Так что при высоких температурах скорость этой реакции практически не зависит от температуры. А для реакции присоединения атома водорода к двойной связи H + C2H4 ® C2H5 энергия активации мала (Eа = 3,4 кДж/моль, так что ее скорость слабо зависит от температуры в широком температурном интервале. И только при температурах намного ниже 0° С начинает сказываться наличие активационного барьера.
Подобных примеров можно привести множество. Очевидно, что правило Вант-Гоффа противоречит не только уравнению Аррениуса, но и многим экспериментальным данным. Откуда же оно взялось и почему нередко выполняется?
Если в приведенном выше математическом выражении для правила Вант-Гоффа подставить вместо скоростей v1 и v2 для данной реакции их зависимости от температуры, в соответствии с уравнением Аррениуса, то после сокращения предэкспоненциальных множителей получим следующее выражение: g = vT +10/vT = е –Е а/R(Т+10)/е –Е а/RТ = е (Еа/R)[1/Т – 1/(T+10)] . Логарифмироване этого уравнения дает: lng = (Eа/R)[1/T – 1/(T + 10)], откуда Еа = Rlng T(T + 10)/10 = 0,83lngT(T + 10). Энергия активации не является функцией температуры, эта зависимость нужна лишь для удобства последующего анализа. Последнее уравнение – это уравнение параболы, в котором физический смысл имеют только положительные значения. Соответствующая диаграмма ограничена двумя ветвями параболы: при g = 2 получаем Еа = 0,58Т(Т + 10), при g = 4 получаем Еа = 1,16Т(Т + 10). К тем же формулам приходим и при использовании десятичных логарифмов. Соответствующие графики двух парабол, для значений g 2 и 4, приведены на рисунке. Их физический смысл заключается в том, что области выполнения правила Вант-Гоффа соответствует только область между параболами. Таким образом, существуют только определенные соотношения между энергией активации реакции и температурой ее проведения, при которых правило Вант-Гоффа выполняется. Ниже нижней ветви температурный коэффициент g 4.
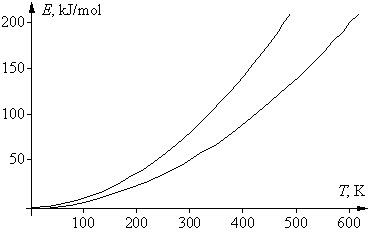
Если посмотреть, какие реакции «укладываются» в указанную довольно узкую область, то окажется, что все эти реакции идут не слишком быстро и не слишком медленно, а с удобной для измерения (при данной температуре) скоростью. Скорость только таких реакций и могли изучать химики во времена Вант-Гоффа. Например, если энергия активации была невелика (меньше 50 кДж/моль), то такая реакция при комнатной температуре заканчивалась за 1–2 секунды; поэтому для изучения ее кинетики следовало значительно понизить температуру, чтобы реакция проходила не быстрее, чем за 10–20 минут. Только в этом случае химики 19 в. успевали отбирать пробы по ходу реакции и анализировать изменение в них концентрации реагентов. Других способов изучения скорости реакции в то время не было. Если это не удавалось (например, водный раствор замерзал), то скорость такой реакции не изучали. Если же энергия активации реакции была велика и при комнатной температуре она шла слишком медленно (многие сутки, или даже недели), то температуру повышали, чтобы реакция шла с удобной для измерения скоростью. И здесь были свои ограничения – например, раствор мог закипеть, т.е. в любом случае исследователи фактически «подстраивали» изучаемую реакцию под область между двумя параболами.
Сейчас химики имеют возможность с помощью различных приборов экспериментально изучать и очень быстрые (идущие в микросекундной области), и очень медленные реакции, для которых температурный коэффициент может быть значительно меньше 2 или значительно больше 4. Поэтому правило Вант-Гоффа, которое, в отличие от уравнения Аррениуса, не имеет четкого физического смысла, представляет лишь чисто исторический интерес и в современной науке не используется. В подавляющем большинстве учебников и монографий по химической кинетике, а также в 5-томной Химической Энциклопедии это правило даже не упоминается. И, тем не менее, если изучаемая реакция идет с удобной для измерения скоростью, например, заканчивается за 30–40 мин, а энергия активации ее еще не измерена, то для предварительной грубой оценки зависимости скорости такой реакции от температуры можно использовать правило Вант-Гоффа. Поэтому это правило приводится во всех школьных учебниках химии.
http://zadachi-po-khimii.ru/obshaya-himiya/skorost-ximicheskoj-reakcii.html
http://www.krugosvet.ru/enc/nauka_i_tehnika/himiya/VANT-GOFFA_PRAVILO.html